
Drug-Induced Liver injury prediction
Many compounds can cause liver injury, after oral administration the first major organ exposed is the liver. The LiverTox is a database of information on the diagnosis, cause, frequency, patterns, and management of liver injury attributable to prescription and nonprescription medications, herbals and dietary supplements.
Checking for the potential to cause liver injury is an important part of the drug discovery process and there are a number of in vitro and in vivo assays that can be used.
High dose studies in safety species are undertaken to identify potential toxicities and to determine safety margins, Clinically, the most relevant reactions include liver necrosis, hepatitis, cholestasis, vascular changes and steatosis. A drug can cause liver toxicity via multiple mechanisms, it can be the result of a direct action of the parent compound or indirectly through reactive metabolites. The drug or its metabolites may cause liver toxicity after specific receptor binding, or reactive metabolites can react with hepatic macromolecules, all leading to direct cytotoxicity. In addition, Immune-mediated idiosyncratic drug reaction has been responsible for numerous serious hepatotoxic events in humans
It would be useful to be able to predict ahead of synthesis whether a molecule was likely to cause liver injury and that is the function of DILIpredictor DOI. Using data form several thousand molecules and a variety of different assays (both in vitro and in vivo) and different species the authors have developed a predictive model. The attraction of this approach is in addition to giving an early flag of potential DILI it also highlights potential species differences and can give an insight into the mechanism.
DILIPredictor required only chemical structures as input for prediction and is publicly available at https://broad.io/DILIPredictor for use via web interface (please don't submit confidential molecules) and with all code available for download from GitHub
https://github.com/srijitseal/DILI_Predictor
I installed like this since it currently does not run with the latest version of python
conda create -n DILIpred python=3.10
conda activate DILIpred
pip install DILIpred
It can then beinstalled as follows.
conda create -n DILIpred python=3.10
conda activate DILIpred
pip install DILIpred
It can then be usef as follows
(DILIpred) chrisswain@Mac-Studio ~ % dilipred -smiles "C[C@@H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C"
If you use DILIPred in your work, please cite: Improved Detection of Drug-Induced Liver Injury by Integrating Predicted In Vivo and In Vitro Data Srijit Seal, Dominic Williams, Layla Hosseini-Gerami, Manas Mahale, Anne E. Carpenter, Ola Spjuth, and Andreas Bender doi: https://doi.org/10.1021/acs.chemrestox.4c00015
100%███████████████████████████████████████████████████████████████ 1/1 [00:01<00:00, 1.14s/it] 2024-07-12 08:29:44.777 | CRITICAL | dilipred.main:predict:458 - The compound is predicted DILI-Positive
The detailed output is contained in a file created.
source,assaytype,description,value,pred,SHAP contribution to Toxicity,SHAP,smiles,smilesr DILI,DILIstFDA,This is the predicted FDA DILIst label,0.8187885151383966,1,N/A,N/A,C[C@@H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C= C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O)c2ccc(F)cc2)C3C)n1 Diverse DILI C,Heterogenous Data ,"Transient liver function abnormalities, adverse hepatic effects",0.7393781727510759,True,Positive,0.003926801602711443,C[C@@H] 1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O )c2ccc(F)cc2)C3C)n1 BESP,Mechanisms of Liver Toxicity,BESP Bile Salt Export Pump Inhibition,0.5655727513227511,True,Positive,0.0003070244326849143,C[C@@H ]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(= O)c2ccc(F)cc2)C3C)n1 Mitotox,Mechanisms of Liver Toxicity,Mitotox ,0.10973983865879627,False,Positive,0.0006130947805277731,C[C@@H]1C2=NN= C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O)c2ccc( F)cc2)C3C)n1 Reactive Metabolite,Mechanisms of Liver Toxicity,Reactive Metabolite Formation,0.19967540492325553,False,Negative,-0.001048102741727997,C[C@@ H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C( =O)c2ccc(F)cc2)C3C)n1 Human hepatotoxicity,Human hepatotoxicity,"Human hepatotoxicity, hepatobiallry",0.7196576912119554,True,Positive,0.007802364907899422,C[C @@H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN( C(=O)c2ccc(F)cc2)C3C)n1 Animal hepatotoxicity A,Animal hepatotoxicity,"Rat, chronic oral administration, Hepatic histopathologic effects, ToxRefDB",0.5867747455286331,True,Positive,0.0030731971130978854,C[C@@H] 1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O )c2ccc(F)cc2)C3C)n1 Animal hepatotoxicity B,Animal hepatotoxicity,"Hepatocellular hypertrophy, rats, ORAD, HESS",0.6646590439473917,True,Positive,0.0013360236463188587,C[C@@H]1C2= NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1 Preclinical hepatotoxicity,Animal hepatotoxicity,"Preclinical hepatotoxicity data from PharmaPendium, Leadscopre, and internal repository with 14- to 28-day rat study data",0.8576928962241468,True,Positive,0.011692057666492625,C[C@@H]1C2= NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1 Diverse DILI A,Heterogenous Data ,Large-scale and diverse ddrug induced liver injury dataset,0.6324274304660036,True,Positive,0.003398315762493277,C[C@@H]1C2 =NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1 source,assaytype,description,value,pred,SHAP contribution to Toxicity,SHAP,smiles,smilesr DILI,DILIstFDA,This is the predicted FDA DILIst label,0.8187885151383966,1,N/A,N/A,C[C@@H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C= C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O)c2ccc(F)cc2)C3C)n1 Diverse DILI C,Heterogenous Data ,"Transient liver function abnormalities, adverse hepatic effects",0.7393781727510759,True,Positive,0.003926801602711443,C[C@@H] 1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O )c2ccc(F)cc2)C3C)n1 BESP,Mechanisms of Liver Toxicity,BESP Bile Salt Export Pump Inhibition,0.5655727513227511,True,Positive,0.0003070244326849143,C[C@@H ]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(= O)c2ccc(F)cc2)C3C)n1 Mitotox,Mechanisms of Liver Toxicity,Mitotox ,0.10973983865879627,False,Positive,0.0006130947805277731,C[C@@H]1C2=NN= C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O)c2ccc( F)cc2)C3C)n1 Reactive Metabolite,Mechanisms of Liver Toxicity,Reactive Metabolite Formation,0.19967540492325553,False,Negative,-0.001048102741727997,C[C@@ H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C( =O)c2ccc(F)cc2)C3C)n1 Human hepatotoxicity,Human hepatotoxicity,"Human hepatotoxicity, hepatobiallry",0.7196576912119554,True,Positive,0.007802364907899422,C[C @@H]1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN( C(=O)c2ccc(F)cc2)C3C)n1 Animal hepatotoxicity A,Animal hepatotoxicity,"Rat, chronic oral administration, Hepatic histopathologic effects, ToxRefDB",0.5867747455286331,True,Positive,0.0030731971130978854,C[C@@H] 1C2=NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O )c2ccc(F)cc2)C3C)n1 Animal hepatotoxicity B,Animal hepatotoxicity,"Hepatocellular hypertrophy, rats, ORAD, HESS",0.6646590439473917,True,Positive,0.0013360236463188587,C[C@@H]1C2= NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1 Preclinical hepatotoxicity,Animal hepatotoxicity,"Preclinical hepatotoxicity data from PharmaPendium, Leadscopre, and internal repository with 14- to 28-day rat study data",0.8576928962241468,True,Positive,0.011692057666492625,C[C@@H]1C2= NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1 Diverse DILI A,Heterogenous Data ,Large-scale and diverse ddrug induced liver injury dataset,0.6324274304660036,True,Positive,0.003398315762493277,C[C@@H]1C2 =NN=C(N2CCN1C(=O)C3=CC=C(C=C3)F)C4=NC(=NS4)C,Cc1nsc(-c2nnc3n2CCN(C(=O) c2ccc(F)cc2)C3C)n1
Overall a useful tool to have to hand.
CCDC: Curated Data Set of Protein Structures
Fantastic news from the Cambridge Crystallographic Data Centre (CCDC), a curated data set of protein structures from the Protein Data Bank (PDB) with predicted hydrogen positions is now available for download. The dataset is taken from the Protein Data Bank (PDB) and has the positions of hydrogens accurately computed, this provides a comprehensive snapshot of protein cavities in the PDB, identifying potential binding sites for small molecules with accurately predicted hydrogen positions for all components.
The news article is here https://www.ccdc.cam.ac.uk/discover/blog/accelerating-drug-discovery-with-the-ccdc-aws-and-intel/.
This large subset of the protein data bank which has be processed using the CCDC's protonation workflow so that reasonable proton positions have been modelled can be downloaded here.
https://www.ccdc.cam.ac.uk/support-and-resources/downloads/.
LLM for Drug Discovery
Whilst general large language models have hit the headlines in recent years, there is a school of thought that smaller domain specific models may actually more useful, in particular in areas like chemistry https://pubs.rsc.org/en/content/articlelanding/2023/dd/d2dd00087c and https://arxiv.org/abs/2402.09391.
A recent preprint describes Tx-LLM a large language model (LLM) for drug discovery https://arxiv.org/pdf/2406.06316. This work from Google Research and Google DeepMind details Tx-LLM, a LLM specifically designed to enhance drug discovery.
Developing therapeutics is a lengthy and expensive process that requires the satisfaction of many different criteria, and AI models capable of expediting the process would be invaluable. However, the majority of current AI approaches address only a narrowly defined set of tasks, often circumscribed within a particular domain. To bridge this gap, we introduce Tx-LLM, a generalist large language model (LLM) fine-tuned from PaLM-2 which encodes knowledge about diverse therapeutic modalities. Tx-LLM is trained using a collection of 709 datasets that target 66 tasks spanning various stages of the drug discovery pipeline. Using a single set of weights, Tx-LLM simultaneously processes a wide variety of chemical or biological entities (small molecules, proteins, nucleic acids, cell lines, diseases) interleaved with free-text, allowing it to predict a broad range of associated properties, achieving competitive with state-of-the-art (SOTA) performance on 43 out of 66 tasks and exceeding SOTA on 22. Among these, Tx-LLM is particularly powerful and exceeds best-in-class performance on average for tasks combining molecular SMILES representations with text such as cell line names or disease names, likely due to context learned during pretraining. We observe evidence of positive transfer between tasks with diverse drug types (e.g., tasks involving small molecules and tasks involving proteins), and we study the impact of model size, domain finetuning, and prompting strategies on performance. We believe Tx-LLM represents an important step towards LLMs encoding biochemical knowledge and could have a future role as an end-to-end tool across the drug discovery development pipeline.
The model was trained using 709 drug discovery datasets comprising 66 tasks formatted for instruction tuning from Therapeutics instruction Tuning (TxT) https://tdcommons.ai collection for tasks across the drug discovery spectrum. These tasks include:
- Evaluating drug efficacy and safety.
- Predicting molecular targets.
- Assessing the ease of manufacturing drugs.
9th Fragment-based Drug Discovery Meeting slides
I'm just back from the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ another fabulous meeting and always great to hear about the multitude of ways that Fragments are impacting drug discovery, from target identification, hit discovery to lead optimisation. A number of people asked if the slides from my talk would be available.
Hopefully this link will be accessible to everyone.
http://cambridgemedchemconsulting.com/news/files/FragHitsMar2024.pdf.
9th Fragment-based Drug Discovery Meeting
I'll be heading over to the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ later today. This event is one of the high points in the Drug Discovery calendar. I'm sure there will be plenty to add to the Fragment-Based Screening section on the Drug Discovery Resources Website.
The aim of the 9th RSC-BMCS Fragment-based Drug Discovery meeting will be to continue the focus on case studies in Fragment-based Drug Discovery that have delivered compounds to late stage medicinal chemistry, preclinical or clinical programmes. The Fragment series was started in 2007 and continues with this theme in having over three-quarters of the presentations focused on case studies. This will be complemented by technology progress in high concentration, NMR, SPR and X-ray screening.
Drug design data sets for testing computational tools
One of the challenges when building novel tools to aid drug discovery is identifying high quality datasets that can be used to test new tools. This is why the D3R datasets are so valuable https://drugdesigndata.org.
These datasets are available from BindingDB and include a variety of important protein targets.
The targets include CathepsinS, BACE1, ABL1) and JAK2.
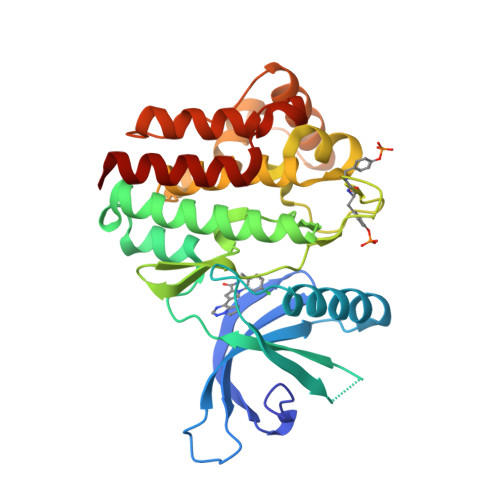
Separation of PK and PD
Just catching up with my reading, I've always been a fan of compounds with slow off-rates and the impact on duration of action.
The situation was elegantly summarised in a publication from earlier in the year from Copeland et al. DOI
A dominant assumption in pharmacology throughout the 20th century has been that in vivo target occupancy-and attendant pharmacodynamics-depends on the systemic concentration of drug relative to the equilibrium dissociation constant for the drug-target complex. In turn, the duration of pharmacodynamics is temporally linked to the systemic pharmacokinetics of the drug. Yet, there are many examples of drugs for which pharmacodynamic effect endures long after the systemic concentration of a drug has waned to (equilibrium) insignificant levels. To reconcile such data, the drug-target residence time model was formulated, positing that it is the lifetime (or residence time) of the binary drug-target complex, and not its equilibrium affinity per se, that determines the extent and duration of drug pharmacodynamics.
I've added it to the page on separation of PK and PD in the Drug Discovery Resources
Updated Drug Discovery Resources
Updated the page on metabolism https://www.cambridgemedchemconsulting.com/resources/ADME/metabolism.html.
And the page on covalent ligands https://www.cambridgemedchemconsulting.com/resources/lead_identification/covalent.html.
Bioisosteres pages updated
I've been updating the Drug Discovery Resources. Over the last few days I've been expanding the section on bioisosteres.
A bioisostere is a molecule resulting from the exchange of an atom or of a group of atoms with an alternative, broadly similar, atom or group of atoms. The objective of a bioisosteric replacement is to create a new molecule with similar biological properties to the parent compound. The bioisosteric replacement may be physicochemically or topologically based. The replacement can attenuate toxicity, modify activity of lead, and/or alter pharmacokinetics or the toxicity of the lead.
Bioisosteres are an essential element in the Medicinal Chemists toolbox and the increasing variety reported is a testimony to the creativity of medicinal chemists.
Annual site review
As 2022 starts I'd like to wish you all a Happy New Year and hope that 2022 marks the start of the recovery from the pandemic.
The Drug Discovery Resources website continues to increase in popularity with 193,322 page views, an increase of 31% over the figure for 2020. The pages were visited by over 95,308 viewers and around 20% of the visitors come back on multiple occasions suggesting they find it useful. The visitors come from 180 different countries with the top countries being
- United States (25%)
- United Kingdom (14%)
- India (13%)
- Germany (3.6%)
- Canada (3%)
- South Korea (2.5%)
Perhaps not unexpectedly one of the popular pages was COVID-19 and the Identification of "Drug Candidates" a checklist for those using virtual screening to identify potential hits for COVID-19 targets.
The other most viewed pages were
- Lipophilicity
- Bioisosteres
- Distribution and Plasma Protein Binding
- Molecular Interactions
- Kinase Inhibitors
- Acid Bioisosteres
- Aromatic Bioisosteres
- Aspartic Acid Proteases
- Solvation and desolvation
- HERG Activity
- Fragment based screening
- ADME
Looking at the operating systems 54% are Windows users, 20% Mac users, 13% Android, 9% iOS and 2% Linux.
I don't know how comprehensive the analytics software is but there is approximately a 50:50 M:F split for Gender.
AI4Proteins videos now online
On June 16/17 2021 RSC CICAG and AI3D held a joint meeting on Protein Structure Prediction. The full lineup of speakers, titles and abstracts can be found here.
Session 1: Session Chair: Professor Jeremy Frey (University of Southampton)
An AI solution to the protein folding problem: what is it, how did it happen, and some implications Professor John Moult (University of Maryland)
Session 2: Session Chair: Dr Melanie Vollmar (Diamond)
So you predicted a protein structure – What now? Dr Thomas Steinbrecher (Schrödinger)
Deep Learning enhanced prediction of protein structure and dynamics Dr Martina Audagnotto (AstraZeneca)
Fireflies-Lévy Flights algorithm for peptides conformational optimization Dr Zied Hosni (University of Sheffield)
Session 3: Session Chair: Dr Chris Swain (Cambridge MedChem Consulting)
How good are protein structure prediction methods at predicting folding pathways? Mr Carlos Outeiral Rubiera (University of Oxford)
Protein-Ligand Structure Prediction for GPCR Drug Design Dr Chris De Graaf (Sosei Heptares)
Session 4: Session Chair: Dr Márton Vass
Using icospherical input data in machine learning on the protein-binding problem Dr Ella Gale (University of Bristol)
Biological sequence design with machine learning Professor Debora Marks (Harvard University)
Session 5: Session Chair: Dr Simone Fulle (Novo Nordisk)
Lessons learned from generative models of biological sequences Professor Aleksej Zelezniak (Chalmers University of Technology)
DeepDock: a deep learning approach to predict ligand binding conformations Dr Oscar Méndez-Lucio (Janssen Pharmaceuticals)
Finding new in silico-based therapeutic strategies for IAHSP Dr Matteo Rossi Sebastiano (University of Turin)
Session 6: Session Chair: Professor Jonathan Goodman (University of Cambridge)
Designing molecular models by machine learning and experimental data Professor Cecilia Clementi (Freie Universität Berlin)
The “almost druggable” genome Professor Tudor Oprea (University of New Mexico)
Session 7: Session Chair: Dr Lucy Colwell (University of Cambridge)
General Effects of AI on Drug Discovery Dr Derek Lowe (Novartis)
Open Access Data: A Cornerstone for Artificial Intelligence Approaches to Protein Structure Prediction Professor Stephen Burley (RCSB PDB, Rutgers University, UCSD)
The videos of the presentations are now available on YouTube and you can access the playlist here https://www.youtube.com/playlist?list=PLBQwbn0mPhvWyTLnN6eFsbIwb5FByrs.
For those wanting a hype free insight into the impact AI might make on Drug Discovery then the presentation by Derek Lowe is well worth watching.
Annual Site Review
As 2021 starts I'd like to wish you all a Happy New Year and hope that 2021 marks the start of the recovery.
The Drug Discovery Resources website continues to increase in popularity with 147,000 page views, an increase of 5% over the figure for 2019. The pages were visited by over 76,000 viewers and around 20% of the visitors come back on multiple occasions suggesting they find it useful. The visitors come from 177 different countries with the top countries being
- United States (28%)
- United Kingdom (14%)
- India (10%)
- Germany (3%)
- Canada (3%)
- Japan (2.5%)
Perhaps not unexpectedly one of the popular pages was COVID-19 and the Identification of "Drug Candidates" a checklist for those using virtual screening to identify potential hits for COVID-19 targets.
The other most viewed pages were
- Lipophilicity
- Calculating Physicochemical Properties
- Bioisosteres
- Distribution and Plasma Protein Binding
- Molecular Interactions
- Kinase Inhibitors
- Acid Bioisosteres
- Aromatic Bioisosteres
- Aspartic Acid Proteases
- Solvation and desolvation
- HERG Activity
- Fragment based screening
- Protacs
Looking at the operating systems 54% are Windows users, 20% Mac users, 12% Android, 10% iOS and 2% Linux.
Cross-referencing the Project Moonshot compounds
The project COVID moonshot is generating a significant amount of data both biochemical data distributed by PostEra and crystallographic data generated and distributed by the team at Diamond.
The COVID Moonshot is an ambitious crowdsourced initiative to accelerate the development of a COVID antiviral. We work in the open with no intellectual property constraints. This way, any scientist can view submitted drug designs and experimental data to inspire new design ideas. We use our cutting-edge machine learning tools and Folding@home's crowdsourced supercomputer to determine which drug designs to send to our partners to make and test in the lab. With each drug design tested, we get closer to our goal.
It is sometimes difficult to cross-reference compounds between multiple sources so I've downloaded the compounds with associated data calculated InChiKeys and then used the InChiKey to link compounds from different sources within Vortex. This means you have the biochemical data together with PDB code (if available) or the fragalysis code for the crystal structure. I've also annotated with identifiers from multiple databases (ChEMBL, PubChem etc.), calculated physicochemical properties (LogP/D, TPSA, HBD/A etc) and then exported in sdf format. I've also clustered the structures to aid navigation.
You can download the zipped sdf file here.
Updated. I was asked if I could provide this file in SMILES format so here it is.
I plan to try and have a look at ways to visualise the data when I can find some free time.
The SARS-CoV-2 main protease as drug target
A very useful primer for those interested in contributing to the ongoing research efforts at COVID moonshot and Open Source COVID-19.
The SARS-CoV-2 main protease as drug target DOI
The unprecedented pandemic of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is threatening global health. The virus emerged in late 2019 and can cause a severe disease associated with significant mortality. Several vaccine development and drug discovery campaigns are underway. The SARS-CoV-2 main protease is considered a promising drug target, as it is dissimilar to human proteases. Sequence and structure of the main protease are closely related to those from other betacoronaviruses, facilitating drug discovery attempts based on previous lead compounds. Covalently binding peptidomimetics and small molecules are investigated. Various compounds show antiviral activity in infected human cells.
Remember a hit in a screen is just the very first step, there is much more to consider before it can be described as a drug candidate.
Drug Discovery Resources Website Stats
I wrote a blog entry about things that should be considered when proposing a hit identified from virtual screening as a drug candidate. Several people have suggested I create an easily identifiable web page so they can reference it. So here it is
COVID-19 and the Identification of "Drug Candidates".
I also thought I'd use the opportunity to look at the Drug Discovery Resources website stats for the first 6 months of 2020.
The Drug Discovery Resources pages are intended to act as a resource for scientists undertaking drug discovery, they were initially based on a course I give but have been expanded to give much more detail and to cover subjects not covered in the course.
The site has been viewed by almost 40,000 viewers with most people viewing a couple of pages per session. The viewers come from over 150 countries, the top countries being.
- United States (28%)
- United Kingdom (16%)
- India (9%)
- Germany (3.5%)
- China (3%)
- Canada (3%)
- Japan (3%)
The most viewed pages were
- Lipophilicity
- Calculating Physicochemical Properties
- Bioisosteres
- Distribution and Plasma Protein Binding
- Molecular Interactions
- Kinase Inhibitors
- Acid Bioisosteres
- Aromatic Bioisosteres
- Aspartic Acid Proteases
- Solvation and desolvation
- Formulation
- Fragment based screening
- Protacs
Looking at the operating systems 55% are Windows users, 20% Mac users, 12% iOS and 12% Android, Chrome dominates the browser stats (64%) with Safari second (17%) and Firefox third (7%).
Another PYMOL session file
A full-length model of glycosylated SARS-Cov-2 spike protein is recently described in literature by Casalino et. al. The PDB files for models are available at https://amarolab.ucsd.edu/covid19.php. These PDB files contain data for spike protein, glycans, lipid membrane, ions, and solvent.
Manish Sud has generated an annotated PyMol session file to view the model of spike protein present in open state conformation. The PyMOL session file is quite helpful during the reading of the article describing the work. It's a bit of elbow grease work to set up appropriate views in PyMOL and Manish has kindly shared it. It's available for download at http://www.mayachemtools.org/Download.html. I'm sure many will find it helpful.
The size of uncompressed PyMOL session file is quite large. It might take few minutes to load it into PyMOL, based on your hardware specifications.
Manish has also provided session files for SARS-CoV-2 Mpro ligands.
More COVID-19 MPro Activity Data
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease.
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
Additional biological results from project moonshot are now available. You can browse the data here https://postera.ai/covid/activity_data.
These results contain a significant milestone with the identification of the first sub micromolar non-covalent inhibitor.
JAG-UCB-a3ef7265-20 has been titrated twice now and has an IC50 of 0.6 uM.
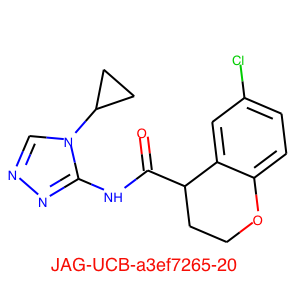
This compound is a racemic mixture and the synthesis of the individual enantiomers is underway, if the activity predominantly lies with a single enantiomer we could see a further improvement in activity. The original submission was based on a pharmacophore search of Enamine based on amino-pyridine hits. I highlight this to underline the importance of simple descriptor-based searches, they are often highly competitive with sophisticated docking studies and require orders of magnitude less compute resources.
Since this research is being conducted in the public domain a number of other people have been able to contribute further ideas based on this exciting discovery.
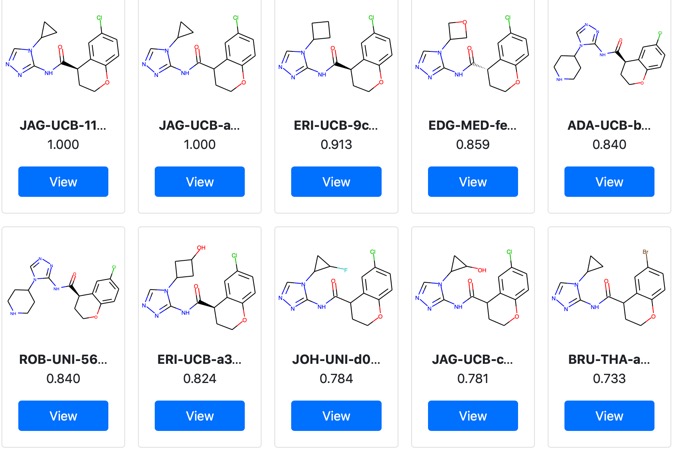
Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease
Full details of the Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease are now published. https://www.biorxiv.org/content/10.1101/2020.05.27.118117v1.full.pdf
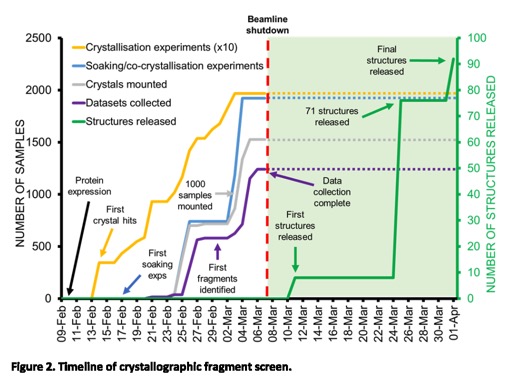
An extraordinary effort highlighted by the timeline shown below.
The results of the first round of biological results from project moonshot are in. You can browse the data here https://postera.ai/covid/activity_data.
A listing of my posts on COVID-19 are here.
First round of MPro bioactivity results
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
The results of the first round of biological results from project moonshot are in. You can browse the data here https://postera.ai/covid/activity_data.
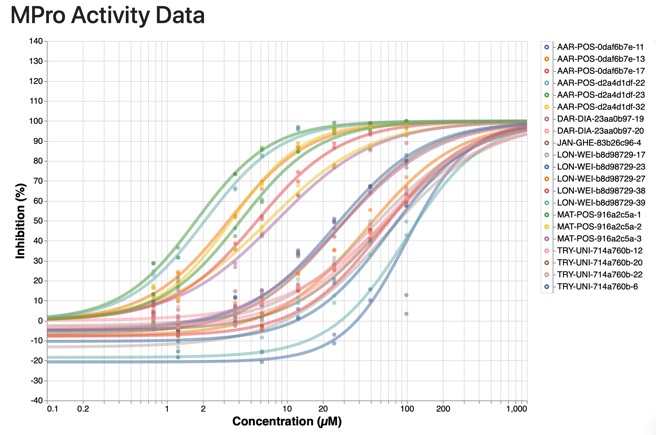
Most of the most active compounds are chloroketones or acrylamides, presumably covalent inhibitors, and they all show selectivity over Trypsin (IC50 >99 uM).
There are a few structures that look more like competitive inhibitors shown below
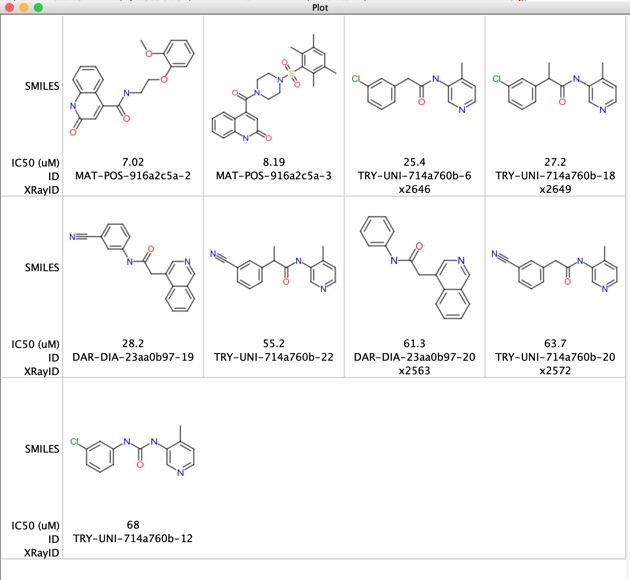
A number of these structures now have crystal structures available.
A sdf file containing these non-covalent structures is here
Fantastic work by all involved.
COVID-19 and the Identification of "Drug Candidates"
One of the really heartening things to come out of the current pandemic is the willingness of many scientists to put aside their own research and throw themselves into the efforts to find a treatment. However, lack of domain expertise is always a problem when scientists enter a new field, so I thought I'd put together a few things to consider.
In silico screening, for docking experiments you need to put considerable effort into ensuring the protein structure used is appropriate, you can't simply download a PDB file from the Protein Data Bank and use it. It will undoubtedly contain errors, you will need check protonation, hydrogen bonds etc. Then there is the issue of deciding which solvent molecules are important. Binding energies, docking scores are not as accurate as many seem to assume and no substitute for an experienced medicinal chemists looking at the bound poses, I've tried to summarise the types of molecular interactions here. Remember to also think about the impact of solvation. For other virtual screening approaches you need to be very careful about the quality of the input data. In many cases it will be heavily biased towards actives.
In silico predictions are no substitute for biological data, if you are using repurposed drugs or available chemicals there is really no excuse for not generating the appropriate in vitro biological data, there are many labs who would be happy to collaborate. If the molecules are novel many custom synthesis companies have offered to help. Remember that the IC50 is probably not that useful, it is likely that you will want to block the target 100% so you need to be above the IC95. In vitro biochemical assays using isolated enzymes will often give a false sense of potency, you should also determine activity in a cell-based assay in the presence of plasma.
If you are proposing a repurposed drug there will be a lot of information about the drug in the public domain, you may well need to search for compound codes, and various drug name synonyms. UniChem is a very useful web service for cross-referencing between chemical structure identifiers.
There are now many free, web-accessible databases some useful starting points are shown in the table below.
| Name | Link | Description |
|---|---|---|
| ChEMBL | https://www.ebi.ac.uk/chembl/ | A database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data). |
| PubChem | https://pubchem.ncbi.nlm.nih.gov | Three linked databases within the NCBI's Entrez information retrieval system. These are PubChem Substance, PubChem Compound, and PubChem BioAssay. Many compounds have links to primary literature and patents |
| Guide to Pharmacology | https://www.guidetopharmacology.org/GRAC/searchPage.jsp | An expert-driven guide to pharmacological targets and the substances that act on them. |
| DrugBank | https://www.drugbank.ca | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug data with comprehensive drug target information |
| NCI Thesaurus | https://ncithesaurus.nci.nih.gov/ncitbrowser/ | NCI Thesaurus (NCIt) provides reference terminology for many NCI and other systems. It covers vocabulary for clinical care, translational and basic research, and public information and administrative activities |
| Clinical Trials | https://clinicaltrials.gov | A database of privately and publicly funded clinical studies conducted around the world |
| FDA | https://www.fda.gov | Food and Drug Administration responsible for safety and efficacy of drugs |
| WIPO | https://www.wipo.int/portal/en/index.html | World IP services |
Find out the original target and mode of action. I've seen a couple of proposed compounds that are known prodrugs, the parent compound is designed to either breakdown or be modified in vivo to yield the active compound. The prodrug may have negligible systemic exposure. Covalent modifiers may look attractive but selectivity is always a concern and they may have narrow therapeutic windows.
Look at the original indication, many anticancer drugs are extremely toxic and could not be given other patients. Similarly, drugs that reduce blood pressure or other physiological changes may be problematic. You may well be able to find counter-screening data, this could highlight problematic off-target activities.
Look at the approved dosing regime, if a drug is only approved for doses of 2 ug/kg there might well be good reasons, and if your proposed drug only has uM activity in the in vitro assays you won't be able to generate sufficient plasma concentrations. Check what safety studies have been undertaken, are they sufficient to support multi-day dosing?
Look at the pharmacokinetics, you should be able to model the dosing regime needed to maintain plasma concentrations above IC95, this will may need to be maintained 24 hours a day. Check protein binding and distribution and use in the predictive modelling.
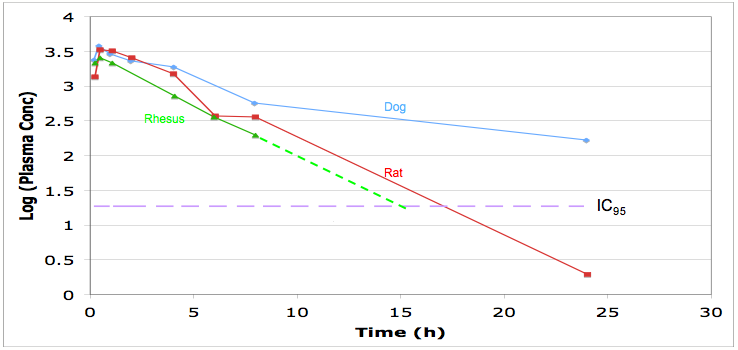
Look for the routes of administration, for in intensive care I suspect many will need the drug to be administered i.v. if there is no intravenous formulation is the drug soluble enough for one to be developed, ber in mind the limitations of intravenous formulations
Many of the patients will be on multiple drugs, both to treat the viral infection but also adventitious bacterial infections and since many are elderly and have pre-existing medical conditions they may have a cocktail of drugs prescribed. Drug-Drug interactions thus become a major concern, any proposed drug to treat the virus that has major interactions with CYP450 enzymes (induction, inhibition or metabolism) is likely to hugely complicate the overall dosing regime.
Check for any toxicity information, particularly black box warnings. HERG inhibition and QT prolongation is an issue that most drug discovery projects have to address at some point. This is particularly worrying if coupled with potential drug-drug interaction described above. You should also be able to find the data from safety studies, these may describe the dose limiting toxicities.
All of this information should be in the public domain, and if you are proposing a compound as a "Drug Candidate" you should not be expecting someone else to pull it all together to decide whether it is worth pursuing clinically.
Updated 26 April 2020
Help design inhibitors of the SARS-CoV-2 main protease
Are you a medicinal chemist currently locked out of your lab?
Why not take a break from writing papers/reports and lend your expertise to this effort, https://covid.postera.ai/covid. They have identified 60 fragment hits and are asking for insight in what should be made next.
We are now asking for your help in designing new inhibitors based on these initial fragment hits: the exceptionally dense readout suggests countless opportunities for growing and merging, and we need many sharp brains to sift through them; it is also what makes us believe that potency can be directly achieved.
The first round of submissions will be reviewed tonight and the selected molecules will be made by Enamine.
Where Drugs Come From: A Comprehensive Look
I was going to highlight this article a while back "Academia and industry: allocating credit for discovery and development of new therapies" DOI but got distracted. However I notice that Derek Lowe has written a commentary that is far more detailed than I could have written on his In the Pipeline Blog.
You can read it here Where Drugs Come From it is well worth spending a little reading, digesting and then sending the link to others.
canSAR BLACK
cansar black v1.1.1 now available - includes improved search, new protein family page, and performance improvements
canSAR is an integrated knowledge-base that brings together multidisciplinary data across biology, chemistry, pharmacology, structural biology, cellular networks and clinical annotations, and applies machine learning approaches to provide drug-discovery useful predictions. canSAR’s goal is to enable cancer translational research and drug discovery through providing this knowledge to researchers from across different disciplines. It provides a single information portal to answer complex multi-disciplinary questions including - among many others: what is known about a protein, in which cancers is it expressed or mutated and what chemical tools and cell line models can be used to experimentally probe its activity? What is known about a drug, its cellular sensitivity profile and what proteins is it known to bind that may explain unusual bioactivity?
Upcoming Conferences
I just thought I'd mention a couple of meetings I'm helping to organise.
2nd RSC-BMCS / RSC-CICAG Artificial Intelligence in Chemistry
Artificial Intelligence is presently experiencing a renaissance in development of new methods and practical applications to ongoing challenges in Chemistry. Following the success of the inaugural “Artificial Intelligence in Chemistry” meeting in 2018 a second meeting has been organised at Fitzwilliam College, Cambridge (2nd to 3rd September 2019). The lineup is now finalised and looks like a great selection of speakers. There is still time to submit posters (closing date 5th July).
Registration is open and there are discounts for RSC members.
The Twitter hashtag - #AIChem19 is already being actively used.
20th SCI/RSC Medicinal Chemistry Symposium
This is Europe’s premier biennial Medicinal Chemistry event, focussing on first disclosures and new strategies in Medicinal Chemistry. It takes place a Churchill College, Cambridge UK, 8 September - 11 September 2019. There is a fantastic lineup of speakers and looks to be one of the highlights of the MedChem calendar. Early career scientists can also take part in a Medicinal chemistry workshop on the Sunday afternoon, a great way for people to learn medicinal chemistry and meet other scientists in a fun and informal setting.
You can register here both RSC and SCI members get a reduced rate, and despite the slightly confusing page on the SCI website you don't have to be a member to attend, just select "Event Member FREE from the dropdown menu and you can register for the event without membership.
Twenty Years of the Rule of Five
It has been over twenty years since Lipinski published his work determining the properties of drug molecules associated with good solubility and permeability. Since then, there have been a number of additions and expansions to these “rules”. There has also been keen interest in the application of these guidelines in the drug discovery process and how these apply to new emerging chemical structures such as macrocycles.
This meeting aims to have a look at the impact the Ro5 has had on drug discovery and as well as looking to the future and how we use these rules in the changing drug compound landscape as drug discovery moves into novel areas of chemistry.
There is a very exciting group of speakers and the timetable has been designed to allow a panel discussion after each session. Given the topic and the speakers I'm sure these will be entertaining sessions.
You can register here and there are discounts for RSC members
Twitter hashtag - #RuleofFive2019
European universities dismal at reporting results of clinical trials
Clinical trial data is some of the most important information in Drug Discovery, after all it is humans we are looking to treat! However analysis of 30 leading institutions found that just 17% of study results had been posted online. The 30 universities surveyed are those that sponsor the most clinical trials in the EU. Fourteen of these institutions had failed to publish a single results summary.
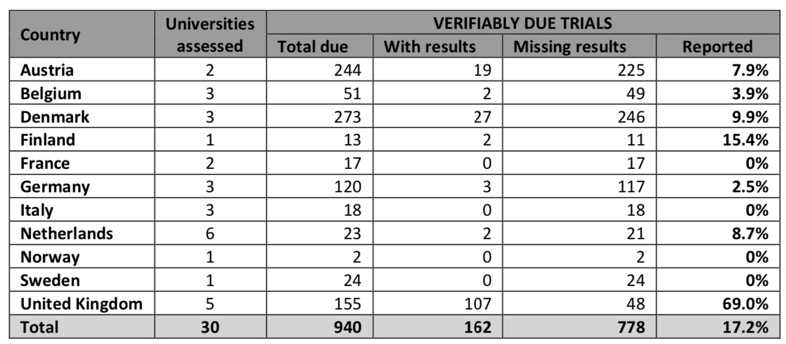
The Universities that have failed to publish a single trial result are highlighted in red in the table below.
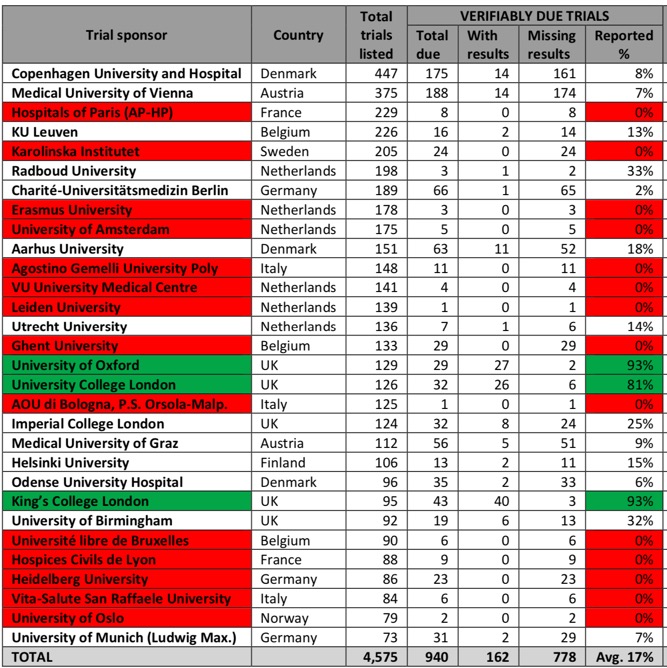
The contrast between the UK universities and the rest of Europe could not be starker,
UK universities in the survey performed significantly better than those in the rest of Europe. The University of Oxford and King’s College London had both published 93% of the trial results due on the register, and University College London had posted 81%.
According to the report every single medical university in the UK is currently working hard to upload missing clinical trial results onto the EU registry, and in many cases onto other registries such as ISRCTN and the US registry Clinicaltrials.gov as well. This demonstrates that where there is a will, there is a way.
If we remove the UK Universities from the analysis the level of reporting falls to a pitiful 7%.
Lack of transparency in clinical trials harms patients. The timely posting of summary results is an ethical and scientific obligation.
Atomwise AIMS awards

I suspect many will have noticed the recent announcement of the Early Results in Drug Discovery Partnership with AI Biotech Company. These are the first results of the Atomwise AIMS awards:
The researchers have been using Atomwise’s AI-powered in silico screening technology to develop therapeutic treatments for, among others, certain types of strokes, hand-foot-and-mouth disease, and an infection that causes reproductive failure in pigs.
The AIMS award program is a great opportunity for university research scientists to easily access AI-assisted structure-based virtual screening technology:
- Customized small molecule virtual screen using AtomNet™ technology
- 72 small molecules predicted to bind to a specific target protein – QC verified by mass spectrophotometry, resuspended and diluted to a convenient concentration, aliquoted into microtiter plates, and delivered at no cost to the researcher
- Support from Atomwise’s medicinal chemists and structural biologists
- Opportunity to receive up to $30K USD to subsidize assay work
If you have a target protein with an X-ray crystal, Cryo-EM, or NMR structure, or with close sequence homology to a protein with available structures, and an assay in place to evaluate 72 potential hits, then you should consider applying.
Full details are on the AIMs awards page and the closing date is 29 April 2019.
How Structural Biologists and the Protein Data Bank Contributed to Recent FDA New Drug Approvals
An interesting review DOI
Discovery and development of 210 new molecular entities (NMEs; new drugs) approved by the US Food and Drug Administration 2010–2016 was facilitated by 3D structural information generated by structural biologists worldwide and distributed on an open-access basis by the PDB. The molecular targets for 94% of these NMEs are known. The PDB archive contains 5,914 structures containing one of the known targets and/or a new drug, providing structural coverage for 88% of the recently approved NMEs across all therapeutic areas. More than half of the 5,914 structures were published and made available by the PDB at no charge, with no restrictions on usage >10 years before drug approval. Citation analyses revealed that these 5,914 PDB structures significantly affected the very large body of publicly funded research reported in publications on the NME targets that motivated biopharmaceutical company investment in discovery and development programs that produced the NMEs.
2019 Medicinal Chemistry Residential School
Registration for the RSC 2019 Medicinal Chemistry Residential School is now open, it takes place in Loughborough, UK 2-7 June 2019.
Through an in-depth programme of lectures, case studies and hands-on tutorial sessions, this five-day course strengthens excellence in medicinal chemistry by advancing understanding of the factors governing modern drug discovery. Full details are here.
Make sure that you register for this course as soon as possible to take full advantage of early bird savings. Registration includes attendance at all sessions, refreshments and lunch on each full day – plus a conference dinner with wine on Thursday 6 June.
ReFRAME library as a comprehensive drug repurposing library
This looks a very interesting resource described in a recent publication. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis DOI.
The ReFRAME collection of 12,000 compounds is a best-in-class drug repurposing library containing nearly all small molecules that have reached clinical development or undergone significant preclinical profiling. The purpose of such a screening collection is to enable rapid testing of compounds with demonstrated safety profiles in new indications, such as neglected or rare diseases, where there is less commercial motivation for expensive research and development.
To date, 12,000 compounds (80% of compounds identified from data mining) have been purchased or synthesized and subsequently plated for screening. In addition, an open-access data portal (https://reframedb.org) has been developed to share ReFRAME screen hits to encourage additional follow-up and maximize the impact of the ReFRAME screening collection.
The website can be searched by structure or text string.
For Example searching for Malaria highlights a number of known therapeutic agents.
https://reframedb.org/#/search?query=malaria&type=string
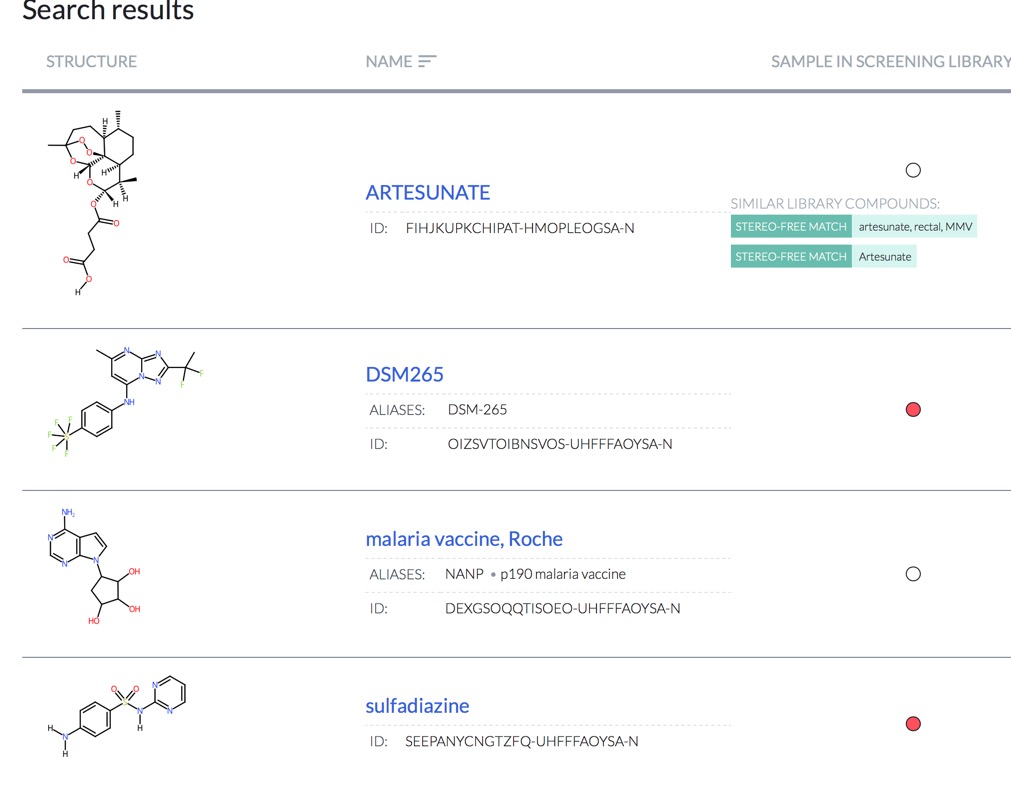
This looks like it will be an invaluable resource.
Properties of clinical candidates
In an excellent publication "Where do recent small molecule clinical development candidates come from?" DOI, the authors give a detailed description on the development of clinical candidates from the initial hit. They also define the changes in physicochemical properties.
An analysis of physicochemical properties on the hit-to-clinical pairs shows an average increase in molecular weight (ΔMW = +85) but no change in lipophilicity (ΔclogP = −0.2), although exceptions are noted. The majority (>50%) of clinical candidates were found to be structurally very different from their starting point and were more complex.
I thought it might be interesting to look at the calculated properties of the 66 clinical candidates. Interestingly many have molecular weights > 500 and only around 30% contain an ionisable group. All structures contain an aromatic ring and 48 molecules contain 3 or more aromatic rings.
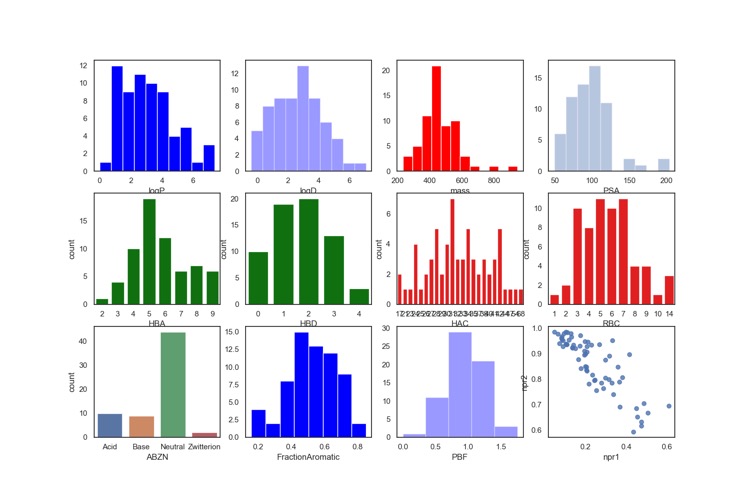
In case anyone was wondering the high molecular weight compounds are not peptides or macrocycles.
The Polypharmacology Browser PPB2
Off-target activity is often ignored and might only be uncovered relatively late in the drug discovery program. Whilst broad spectrum screening is available it can be rather expensive. Predicting potential off-target activities is an attractive approach and this paper describes the development of a prediction tool using nearest neighbours combined with machine learning.
The Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning DOI
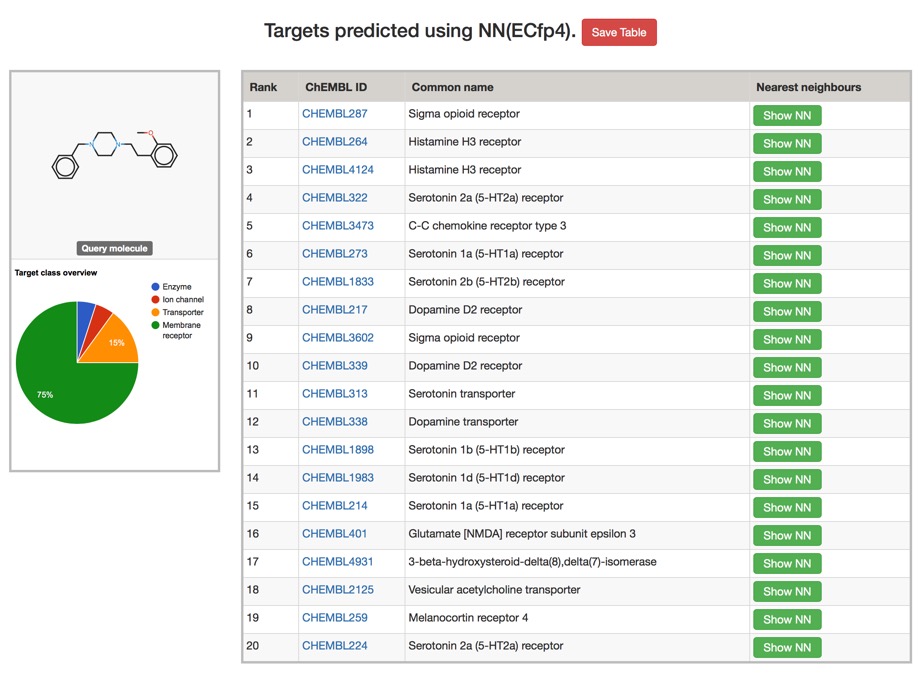
To build PPB2 we collected a bioactivity dataset of all compounds having at least IC50 < 10 uM on a single protein target in ChEMBL22 considering only high confidence data points as annotated in ChEMBL and only targets for which at least 10 compounds were documented
You can try it out here PPB2., depending on the model chosen the results are calculated in a couple of minutes, but don't post your proprietary molecules. Typical results are shown below, clicking on the green "Show NN" button shows the most similar structures.
How reliable is the literature?
In the past I've mentioned some of the concerns about antibody selectivity, the problems with irreproducible studies and the need for well characterised chemical probes. Elisabeth Bik has been looking at concerns with some of the images in the published literature, The prevalence of inappropriate image duplication in biomedical research publications. mBio 7(3):e00809-16. DOI. her Twitter feed contains yet more examples from the current literature, well worth a browse.
So completing this set, I looked at 101 papers, all published in the same month in the same journal. Of these, 63 contained photographic images (the others had only line graphs and/or tables). Of those, 8 have potential duplications. That is 12.7%
With all the advances in AI and image recognition I'm slightly disappointed that there is not a programme that can do this automatically for Elisabeth.
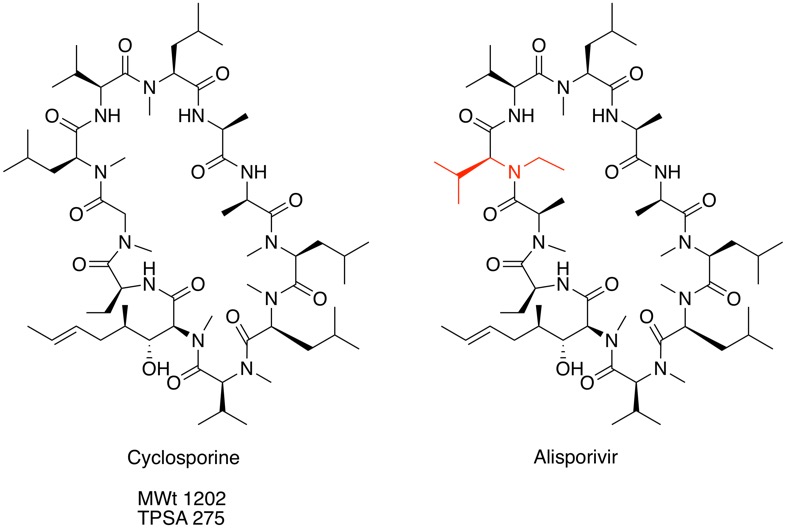
Macrocycles in Drug Discovery
I've created a new page in the Drug Discovery Resources section describing the use of macrocycles in drug discovery. With the advent of more challenging drug discovery targets such as protein-protein interactions there is renewed interest in molecules that are beyond the "Rule of 5". Macrocycles despite apparently undesirable physicochemical properties (High MWt, polar surface area etc.) can have good cell penetration and oral bioavailability.
There is also an upcoming meeting on Macrocycles, 3rd RSC BMCS Medicinal Chemistry Symposium on Macrocycles. Monday-Tuesday, 8th-9th October 2018, GlaxoSmithKline, Stevenage, UK. Full details are here http://www.maggichurchouseevents.co.uk/bmcs/Macrocycles-2018.htm. #BMCS_Macrocycles
The objective of this symposium is to promote scientific interaction between scientists with a shared interest in the field of Macrocycles. This area is responsible for a growing number of therapeutic approaches and development candidates, all of which go ‘beyond the rule-of–five’. As a researcher in this field, come along to hear about the latest advances and also to share in some of the secrets of discovering therapeutic agents which go beyond Lipinski’s rules.
Pitfalls to avoid when building a Computational Therapeutics Company
Everyday I seem to hear about another tech company moving into healthcare, whilst I'm certain that Artificial Intelligence and Machine Learning has the potential to make a significant impact this advice should be compulsory reading for all involved.
Updated bioisosteres pages
Bioisosteres are an essential tool in the medicinal chemistry toolkit.
Bioisosteres are chemical substituents or groups with similar steric or electrostatic properties which produce broadly similar biological properties to another chemical compound.
Bioisosteres won't always give improved properties, and sometimes we find that a transformation that improves metabolic stability in one series might have the reverse effect in another. However they provide useful (literature precedented) starting points for structural transformations that allow exploration of structure activity relationships. Sometimes they yield similar activity but offer alternative structural vectors for exploration, sometimes they simply improve solubility.
I've now updated the bioisosteres section of the Drug Discovery Resources including new examples people have kindly sent to me.
Computational Tools Updated
I've updated the computational tools page in the Drug Discovery Resources.
First Disclosures at ACS San Francisco
The ACS session organised by Division of Medicinal Chemistry includes a day of first disclosures of potential clinical candidates for the first time. Beth Halford was there and tweeted a series of hands drawn structures as they were disclosed. I've redrawn the structures and converted them into a single sdf file. I've also used Jupyter Notebook to calculate the physicochemical properties and plot them as shown in the image below.
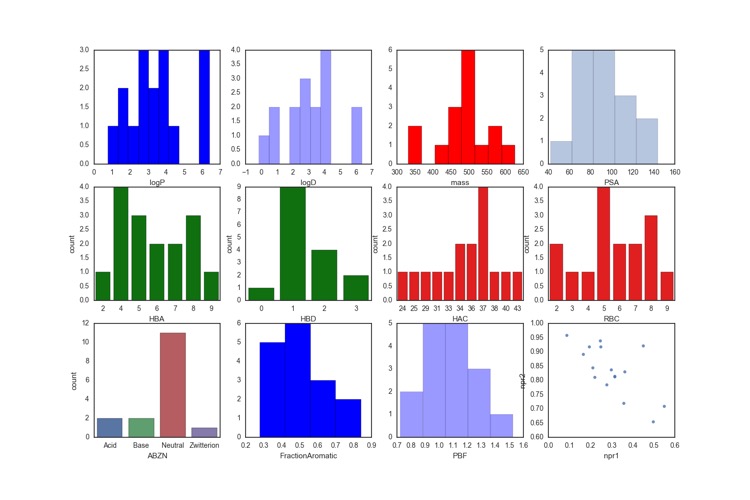
As can be seen, many of the compounds are on the large size with molecular weights >450, a third of them have ionisable groups which serves to bring down the calculated LogD. All molecules contain an aromatic ring, indeed many contain multiple rings. Interestingly we see a number of examples where a biphenyl ring system bears multiple ortho substituents presumably locking the two aryl rings orthogonal, no mention of atrope isomers though. This might also reduce the planarity and increase the 3D shape.
The sdf file is available here FirstDisclosures.sdf.zip if anyone spots any errors in the structures please let me know.
Drug Discovery Resources Updates
I've been updating the Drug Discovery Resources section.
In particular I've added a section on target prediction tools, updated the section on Analysis of HTS data, and expanded the section on aggregation in bioassays to include a recent publication giving an example of target specific aggregation.
The crystal structure showing the interaction is available (PDB 5MU8) and is displayed below using 3Dmol.js, the ligand (JNJ525) is shown in red..
Mouse Controls
| Movement | Mouse Input | Touch Input | ||
|---|---|---|---|---|
| Rotation | Primary Mouse Button | Single touch | ||
| Translation | Middle Mouse Button or Ctrl+Primary | Triple touch | ||
| Zoom | Scroll Wheel or Second Mouse Button or Shift+Primary | Pinch (double touch) | ||
| Slab | Ctrl+Second | Not Available |
In addition I've updated the section on molecular interactions.
The BMCS Mastering MedChem III: 3rd RSC-BMCS symposium on mastering medicinal chemistry
This meeting is aimed at all those who wish to become better drug hunters and heed warnings from the past. (22 March 2017 09:00-19:00, Cardiff Bay, United Kingdom).
In the main there are two types of drug discovery programmes: those that hit serious problems and those that are going to hit serious problems. The difference between success and failure is how we, as medicinal chemists, tackle and resolve the problems
Sounds a great meeting both for those starting out in their careers and for those looking to pick up new tips.
Drug Discovery Resources Site Update
I've completed a few updates to the Drug Discovery Resources website, this includes fixing any broken links that people have mentioned to me, and also starting a new section in the Pre-clinical toxicity on Mutagenicity.
Cheminformatics for Drug Design: Data, Models & Tools
Still a few places left at the Cheminformatics for Drug Design: Data, Models & Tools meeting organised by SCI's Fine Chemicals Group and RSC's Chemical Information and Computer Applications Group has been extended.
Imperial War Museum, Duxford, UK Wednesday 12 October 2016
Full details are available here https://www.soci.org/Events/Display-Event?EventCode=FCHEM481
Sounds an excellent meeting and you will have a chance to look around the aircraft at the Duxford Imperial War Museum.
Target Valaidation site update
TargetValidation.org has been updated.
This release brings new web displays and plenty of extra data to assist you in drug discovery and validation:
- 30,591 targets
- 9,425 diseases
- 4.8 million evidence
- 2.4 million target-disease associations
There are also new Web Widgets for both 'RNA baseline expression' and 'Protein Structure' of a target. In the latter, you can now rotate the protein structure, change its colour, zoom in and out, and highlight any amino acid residue:
More information is available on the blog
Cheminformatics for Drug Design: Data, Models & Tools
I’ve just heard that the poster deadline for the Cheminformatics for Drug Design: Data, Models & Tools meeting organised by SCI's Fine Chemicals Group and RSC's Chemical Information and Computer Applications Group has been extended.
Imperial War Museum, Duxford, UK Wednesday 12 October 2016
Full details are available here https://www.soci.org/Events/Display-Event?EventCode=FCHEM481
Sounds an excellent meeting and you will have a chance to look around the aircraft at the Duxford Imperial War Museum.
19th RSC/SCI Medicinal Chemistry Symposium
I’m delighted to highlight the first announcement of the 19th RSC/SCI Medicinal Chemistry Symposium to be held in Cambridge in September 2017. Europe’s premier biennial Medicinal Chemistry event, focusing on first disclosures and new strategies in medicinal chemistry.

Solutions for Drug-Resistant Infections Meeting
SDRI 2017 is a multi-disciplinary scientific conference for the Asia Pacific region focused on Solutions for Drug Resistant Infections. This inaugural conference theme is New Drugs for Drug-Resistant Infections. The conference will take place at the Brisbane Convention and Exhibition Centre in Australia from 3 - 5 April, 2017.
The program is expected to attract 400 international participants and will provide a fantastic forum for researchers and industry representatives working in the space of microbiology, virology, parasitology, genomics, pharmacology and medicinal chemistry, to network and discuss new ways to solve the global challenge of drug-resistant infections. Our goal for SDRI 2017 is to lead a concerted discussion to set three priorities and guide research efforts towards global solutions for drug resistance research.
Conference themes:
- Antimicrobial drug discovery
- Improvements to existing anti-infective agents and repurposing
- New Drug Targets
- Alternate therapies
- Navigating the pipeline
- International Models and Funding
- Vector control and vaccines
International keynote speakers confirmed:
- Professor Dame Sally Davies DBE FMedSci FRS, Chief Medical Officer for England
- Professor Ramanan Laxminarayan, Director for Center for Disease Dynamics, Economics & Policy (CDDEP), Washington and Vice-President for Research & Policy at Public Health Foundation of India (PHFI)
CO-ADD web portal
The CO-ADD web portal is now live You can now submit your free antimicrobial screening request, download all forms and access your primary screening, cytotoxicity, hit confirmation and hit validation reports online on the new secure CO-ADD user portal.
CO-ADD (Community for Open Antimicrobial Drug Discovery) is a not-for-profit initiative led by academics at The University of Queensland. Our goal is to screen compounds for antimicrobial activity for academic research groups for free. We aim to help researchers worldwide to find new, diverse compounds to combat drug-resistant infections.
Chemical Probes Portal Updated
The Chemical Probes Portal has been updated, the new site includes a lot more data about the existing probes, reviewer ratings and their comments.
A chemical probe is simply a reagent—a selective small-molecule modulator of a protein’s function—that allows the user to ask mechanistic and phenotypic questions about its molecular target in cell-based and/or animal studies. These are tools not drugs, they allow scientists to investigate the relationship between a molecular target and the broader biological consequences of modulating that target in cells or organisms. In general the focus is on specificity for the target rather than pharmacokinetics.
Community for Open Antimicrobial Drug Discovery
A little while ago I mentioned The Community for Open Antimicrobial Drug Discovery effort to provide free compound screening against a variety of infective agents. .
Primary Screening of a 1mg sample Test against key ESKAPE pathogens, E. coli, K. pneumoniae, A. baumannii, P. aeruginosa, S. aureus (MRSA), as well as the fungi C. neoformans and C. albicans, at a single concentration.
Hit Confirmation:- Confirm activity with minimum inhibitory concentration and counterscreen for cytotoxicity and membrane interaction.
Hit Validation:- Test the positive hit against a broader panel of microbes and evaluate the basic drug qualities of actives. CO-ADD will screen your compounds for free and make no claim to IP. The linked flyer gives full details
You can also read more details in this Nature article DOI
It looks like they have achieved an important milestone!
Thanks to our research community we have received 100,000 compounds from 30 countries in 15 months! Make a difference: clear the fridge, empty the shelves and send through your compounds for free antimicrobial screening against 5 bacteria and 2 fungi. We would also like to acknowledge the contribution of the French National Chemical Library that has safely arrived in Brisbane last week!
So if you have compounds sitting in the back of cupboards why not send them to be tested
Open Targets
A little while back I mentioned the Centre for Therapeutic Target Validation, well it seems that it has now been renamed Open Targets.
The Target Validation platform brings together information on the relationships between potential drug targets and diseases. The core concept is to identify evidence of an association between a target and disease from various data types. A target can be a protein, protein complex or RNA molecule, but we integrate evidence through the gene that codes for the target. In the same way, we describe diseases through a structure of relationships called the Experimental Factor Ontology (EFO) that allows us to bring together evidence across different but related diseases.
There is a video online describing it in more details https://vimeo.com/149309356
This is an absolutely invaluable resource for anyone involved in drug discovery, simply type your query into the text box and submit the query.

This update also bring programmatic access to the data via a series of REST services, the API is fully documented. All the methods are available via a GET request and will serve the output formatted as json. There is a getting started tutorial available.
Unlocking Finance for Drug Discovery
This looks like an interesting meeting for those looking for funding drug discovery
This event is aimed at early and mid-career scientists who are looking to learn more about funding early stage drug discovery or how to attract follow-on investment. A panel of experts, representing several investors from the industry and charities will share their experience and engage in a discussion on the future of drug discovery landscape. Find out about what propels a drug discovery project to secure funding and investment
Location: Cancer Research UK Cambridge Institute Lecture Theatre, Cambridge Biomedical Campus, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE
Date:21 April 2016
Medicines for Malaria Venture 14th call for proposals
MMV have just announced a call for proposals in the following three areas:
Compounds addressing the key priorities of the malaria eradication agenda. Novel families of molecules in the hit-to-lead or lead optimization stages are sought without G6PD deficiency liabilities that either kill or reactivate hypnozoites for use as part of a P. vivax radical cure; or have dual activity against asexual and sexual stages (gametocytes) for treatment and transmission blocking.
Asexual liver and blood stages. Novel chemical series with EC50<500nM and which have one or more of the following key features: A novel mechanism of action A long half-life (ideally >4h in rodents) and confirmed in vivo efficacy. For advanced series, we are seeking novel compounds with, ideally, a predicted human half-life >100h and a predicted single human dose <500mg or three day human dose of <50 mg.
Novel approaches for screening. To help identify new phenotypic and/ or target based hits, as well as confirm activity of MMV compounds on all human malaria asexual blood stages, new screening proposals are sought.
They have also published Target product profiles & target candidate profiles.
Web browsers used in Drug Discovery
Last week I posted this observation
More and more of the companies/groups that I'm working with are moving away from desktop applications to providing a web-based portfolio of applications for drug discovery. Most seem to use a combination of commercial tools with a selection of in house apps. Whilst this has many advantages it does raise the question about which web browser should they support? Whilst NetMarketshare still has Internet Explorer at 44% this is probably not a good metric to measure browser usage in the Drug Discovery Sector. So for the last couple of months I've been monitoring the web browsers used to access the Drug Discovery Resources since it is unlikely that anyone not interested in drug discovery would spend much time browsing these pages. The results are interesting.
The ranking since 1 Jan 2016 to date is
- Chrome 55%
- Safari 20%
- Firefox 16%
- Internet Explorer 4%
Looking at operating systems
- Windows 57%
- Macintosh 23%
- iOS 11%
- Android 8%
So the lack users of Internet Explorer is not due to the absence of Windows users. This must have implications for all developers, the users appeared to have moved to the more modern web browsers.
Update
I've now data from around 10 different sites involved in drug discovery or software/databases to support drug discovery, ranging from small sites with about 10,000 hits a month to major sites with many millions of hits a month, and I've now included the average data in the table below.
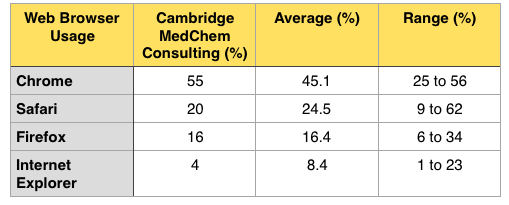
It looks like the data from Drug Discovery Resources reasonably reflects the usage in the Drug Discovery sector.
Web-based tools
More and more of the companies/groups that I'm working with are moving away from desktop applications to providing a web-based portfolio of applications for drug discovery. Most seem to use a combination of commercial tools with a selection of in house apps. Whilst this has many advantages it does raise the question about which web browser should they support? Whilst NetMarketshare still has Internet Explorer at 44% this is probably not a good metric to measure browser usage in the Drug Discovery Sector.
So for the last couple of months I've been monitoring the web browsers used to access the Drug Discovery Resources since it is unlikely that anyone not interested in drug discovery would spend much time browsing these pages. The results are interesting.
The ranking since 1 Jan 2016 to date is
- Chrome 56%
- Safari 20%
- Firefox 16%
- Internet Explorer 4%
Looking at operating systems
- Windows 57%
- Macintosh 23%
- iOS 11%
- Android 8%
So the lack users of Internet Explorer is not due to the absence of Windows users. This must have implications for all developers, the users appeared to have moved to the more modern web browsers.
Update
A number of readers/companies have contacted me since I published with broadly similar results, I hope to compile and publish the anonymised results next week.
Global Health Compound Design Webinar - recording & next meetings
During 2016 Global Health are running a series of webinars on the subject of compound design. The programme for future meetings is available below (the agenda will develop through the year).
Date Agenda (& timing of each item in the recording when available)
21st Jan 2016
Introduction to meetings, Mark Gardner Application of PK Tools in the optimisation of a series for the treatment of leishmaniasis, Gavin Whitlock, Sandexis, working with DNDi Hints and tips to working with DataWarrior, Isabelle Giraud, Actelion, slides Isabelle Giraud, DataWarrior demonstration" Recording
25th Feb 2016
Visceral leishmaniasis TCP & screen sequence, Charlie Mowbrary, DNDi Malaria Target Candidate Profiles, stage gates and implications for successful malaria drug discovery, Paul Willis, MMV Registration
17th Mar 2016 "DataWarrior advanced data analysis, Isabelle Giraud, Actelion Using the RSC Medicinal Chemistry Toolkit in Drug Discovery Projects, Andy Davis, AZ The RSC Medicinal Chemistry Toolkit is a free suite of resources to support the day-to-day work of drug discovery scientists. It was developed to provide difficult-to-access, but industry-validated tools in a portable format. The presentation will show with worked examples, how the RSC Medicinal Chemistry Toolkit (Apple only) can be used to support design strategy thinking and structure-activity optimization. https://itunes.apple.com/gb/app/medicinal-chemistry-toolkit/id910073742?mt=8" Registration
21st Apr 2016
Free data pipelining tool KNIME in compound design & analysis Introduction to KNIME & use cases in drug discovery – further details tbd Registration
There are more details here.
Free webinar to discuss compound design.
Webinar to discuss compound design. This meeting:
* Brief introduction - Mark Gardner (AMG Consultants)
* Application of PK Tools in the optimisation of a series for the treatment of leishmaniasis, Gavin Whitlock, Sandexis, working with DNDi.
* Hints and tips to working with DataWarrior, Isabelle Giraud, Actelion
Register here https://attendee.gotowebinar.com/register/6743114969530154241
Insects in Drug Discovery
The company N2MO offers the use of insects as model organisms. They can be used for ADME screening in particular brain penetration studies.
The Grasshopper: A Novel Model for Assessing Vertebrate Brain UptakeOlga Andersson, Steen Honoré Hansen, Karin Hellman, Line Rørbæk Olsen, Gunnar Andersson, Lassina Badolo, Niels Svenstrup, and Peter Aadal Nielsen EntomoPharm R&D, Medicon Village, Lund, Sweden (O.A., K.H., G.A., P.A.N.); Department of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark (S.H.H., L.R.O.); and Division of Discovery Chemistry and Drug Metabolism and Pharmacokinetics, H. Lundbeck A/S, Copenhagen, Denmark (L.B., N.S.) Received April 10, 2013; accepted May 10, 2013
ABSTRACT The aim of the present study was to develop a blood-brain barrier (BBB) permeability model that is applicable in the drug discovery phase. The BBB ensures proper neural function, but it restricts many drugs from entering the brain, and this complicates the development of new drugs against central nervous system diseases. Many in vitro models have been developed to predict BBB permeability, but the permeability characteristics of the human BBB are notoriously complex and hard to predict.
Consequently, one single suitable BBB permeability screening model, which is generally applicable in the early drug discovery phase, does not yet exist. A new refined ex vivo insect-based BBB screening model that uses an intact, viable whole brain under controlled in vitro-like exposure conditions is presented.
This model uses intact brains from desert locusts, which are placed in a well containing the compound solubilized in an insect buffer. After a limited time, the brain is removed and the compound concentration in the brain is measured by conventional liquid chromatography-mass spectrometry. The data presented here include 25 known drugs, and the data show that the ex vivo insect model can be used to measure the brain uptake over the hemolymph-brain barrier of drugs and that the brain uptake shows linear correlation with in situ perfusion data obtainedinvertebrates.Moreover,this study shows that the insect ex vivo model is able to identify P-glycoprotein (Pgp) substrates, and the model allows differentiation between low-permeability compounds and compounds that are Pgp substrates.
Hepatotoxicity
I've expended the preclinical toxicity section to include a page on hepatotoxicity. This gives some details of the common assays and markers used to evaluate the potential hepatotoxicity.
Time Dependent Inhibition
I've just updated the Drug Discovery Resources page on CYP Interactions, included a section on Time Dependent Inhibition (TDI).
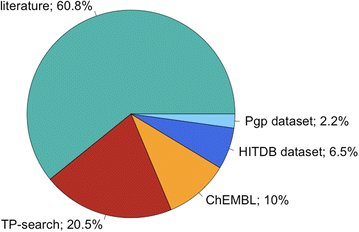
Metrabase
p>The Metabolism and Transport Database (Metrabase) is a cheminformatics and bioinformatics resource that contains curated data related to human small molecule metabolism and transport, Journal of Cheminformatics 2015, 7:31 DOI. Currently it includes interaction data on 20 transporters, 3438 molecules and 11649 interaction records manually abstracted from 1211 literature references and supplemented with data from other resources as shown in the image below taken from the original publication.
I've added this and more details to the Transporters page of the Drug Discovery Resources
Grant Funding Research
I've just updated the page listing possible sources of grant funding for drug discovery research. In particular I've extended the listing of disease specific resources, these may be particularly useful for rare or neglected diseases.
Lack of reproducibility with antibodies
A slightly worrying article in Nature, Reproducibility crisis: Blame it on the antibodies.
The lack of reproducibility of published data on potential drug targets has been highlighted on several occasions DOI and it has been suggested that this is a major factor in the failure rate for phase 2 clinical trials DOI.
In almost two-thirds of the projects, there were inconsistencies between published data and in-house data that either considerably prolonged the duration of the target validation process or, in most cases, resulted in termination of the projects.
Antibodies have rapidly become a key tool in understanding and identifying new drug targets and potentially used as biomarkers to identify patients. However it is clear that many of the 2 million commercially available antibodies need to be checked rigorously, with some scientists claiming more than half are unreliable.
In 2011, an evaluation4 of 246 antibodies used in epigenetic studies found that one-quarter failed tests for specificity, meaning that they often bound to more than one target. Four antibodies were perfectly specific — but to the wrong target.
Caveat emptor.
Free Compound Screening for antimicrobial activity
The concerns about antibiotic resistance are well known and indeed have made headlines in the mainstream press. Here is a chance to help find the next generation of antibiotics.
Do you have interesting compounds sitting on the shelf? Perhaps you would be interested in having them screened for antibiotic activity for free?
The Community for Open Antimicrobial Drug Discovery would like to hear from you, their goal is to screen compounds from academic research groups from anywhere in the world for free.
The requirements are pretty minimal
We ask for 1-2 mg of pure compound which will be used for primary screening, hit confirmation, and if active will be used for a broader antimicrobial screening, cytotoxicity and a check for its purity. We require all compounds to be soluble in water or DMSO and to be shipped as dry material in appropriate containers, such as 1-2 mL Eppendorf tubes. For larger collections we can arrange plates or tube-racks.
In the primary screen they test against against key ESKAPE pathogens, E. coli, K. pneumoniae, A. baumannii, P. aeruginosa, S. aureus (MRSA), as well as the fungi C. neoformans and C. albicans. The ‘ESKAPE’ pathogens that are responsible for two-thirds of all health care-associated infections and resistant strains of these bacteria represent the greatest unmet need in antibacterial drug development.
Annual Site Review
At the end of each year I take the opportunity to look at the website analytics to see what parts of the website are the most popular. Overall there was a 15% increase in the number of page views up to 75,000. Average time on a page was 2 mins suggesting the content is engaging with the viewers.
Nine of the top ten most popular pages were from the Drug Discovery Resources Pages which I am delighted to see, since it suggests that the work entailed in putting the resources together is worthwhile.
The most viewed pages were
Drug versus Metabolite similarity
A recent paper from Douglas Kell et al DOI has provoked much discussion, especially since it was highlighted on In the Pipeline. The authors suggest that similarity to a human metabolite may be a useful as an indication of how “drug like” a molecule might be.
We exploit the recent availability of a community reconstruction of the human metabolic network (‘Recon2’) to study how close in structural terms are marketed drugs to the nearest known metabolite(s) that Recon2 contains. While other encodings using different kinds of chemical fingerprints give greater differences, we find using the 166 Public MDL Molecular Access (MACCS) keys that 90 % of marketed drugs have a Tanimoto similarity of more than 0.5 to the (structurally) ‘nearest’ human metabolite. This suggests a ‘rule of 0.5’ mnemonic for assessing the metabolite-like properties that characterise successful, marketed drugs. Multiobjective clustering leads to a similar conclusion, while artificial (synthetic) structures are seen to be less human-metabolite-like. This ‘rule of 0.5’ may have considerable predictive value in chemical biology and drug discovery, and may represent a powerful filter for decision making processes.
Whilst this represents an interesting observation I was rather concerned about the choice of a Tanimoto coefficient of 0.5, and decided to repeat the analysis.
The recon-2 dataset was downloaded as a Matlab file, this was exported as a plain text file and Rajarshi Guha converted them to SMILES strings and removed duplicates (and did a comparison with PAINS). I imported these structures into a MOE database and then used a SVL script to compare the recon2 with several other datasets. This included DrugBank that includes details of just under 7000 drug entries, a cleaned up subset of leadlike molecules from Zinc, and BindingDB a public, web-accessible database of measured binding affinities I downloaded in 2008. The datasets were first compared to each other using the MACCS fingerprints with a Tanimoto cutoff of 0.5.
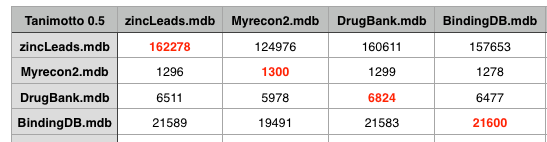
As the table above shows using a Tanimoto coefficient of 0.5 indeed 90% of the molecules in DrugBank are similar to a molecule in recon2, however the same is true for Zinc and BindingDB, indeed at a Tanimoto coefficient of 0.5 all the datasets are pretty similar.
If we increase the Tanimoto coefficient to 0.85 we start to see some resolution, recon2 looks to have more overlap with DrugBank than with either Zinc or BindingDB. However this may simply be a reflection of the fact that DrugBank contains a significant proportion of natural product derived compounds.
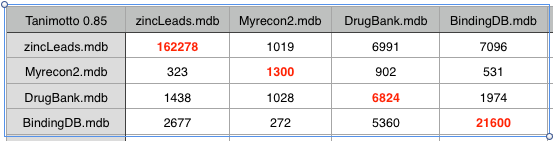
The key question of course is “Does this help us to identify compounds that are likely to fail in development?”. It would be really useful to compare with successful drugs and those that fail in development however I’m not aware of any dataset of of failed drug candidates (if anyone knows of one please let me know). However to in an effort to perhaps get some insight I’ve compared the recon2 set with a dataset of drugs that have been withdrawn (for a variety of reasons). As might be expected using a Tanimoto coefficient of 0.5 offers little discrimination. Increasing to 0.85 it looks like there might be a signal there, but the dataset is too small for firm conclusions.
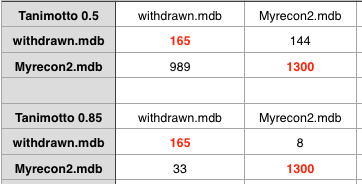
In summary, this limited exploration suggests there may be something worth following up, but that a Tanimoto of 0.5 simply offers little discrimination.
Seven pharma companies provide access to stalled development compounds
UK researchers will be granted access to a ‘virtual library’ of deprioritised pharmaceutical compounds through a new partnership between the Medical Research Council (MRC) and seven global drug companies, announced today by Business Secretary Vince Cable.
AstraZeneca, GlaxoSmithKline, Janssen Research & Development LLC*, Lilly, Pfizer, Takeda and UCB will each offer up a number of their deprioritised molecules for use in new studies to improve our understanding of a range of diseases. A full list of available compounds will be published later this year, when UK scientists will be able to apply for MRC funding to use them in academic research projects.
This has the potential to a really exciting resource for scientists to explore the pathways involved in a variety of different diseases, and since the compounds have apparently undergone some development it may provide a boon to those involved in repurposing drugs. Much will of course depend on the compounds offered but perhaps other companies will follow suit.
Drug Discovery Resources Updates
I’ve updated the Drug Discovery Resources, in particular I’ve updated the section on Brain Penetration to include more on predictive models.
I’ve also updated the page on Grant Funding Research.
Worth a look.
The a third edition of the popular book, The Organic Chemistry of Drug Design and Drug Action by Silverman and Holladay has just been released, I’ve added it to the book list.
Vortex users might be interested in a new script that implements an interesting paper from Wagner et al Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties DOI that describes an algorithm to score compounds with respect to CNS penetration.
Lilly MedChem rules can now be installed using Homebrew. In late 2012 Robert Bruns and Ian Watson published a paper entitled Rules for Identifying Potentially Reactive or Promiscuous Compounds DOI. This article describes a set of 275 rules, developed over an 18-year period, used to identify compounds that may interfere with biological assays, allowing their removal from screening sets.
Drug Discovery Resources Updates
I’ve made a couple of updates to the Drug Discovery Resources pages. In particular I’ve updated the Published fragments Hits to include more examples, details of “promiscuous” compounds and summary of detection technologies and the targets explored. I’ve also updated the Aspartic Protease inhibitors page.
As ever comments and/or suggestions very welcome.
Bringing Open Source to Drug Discovery demo
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. I’ve now recorded the demo I showed and put it on YouTube
https://www.youtube.com/watch?v=sG9vDIfp0NE&feature=youtu.be
If you want any further information I’d be happy to try and help.
Bringing Open Source to Drug Discovery
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. As I promised at the meeting I’ve published the slide deck that now includes 25 pages on links and resources that I hope you will find useful.
Bringing Open Source to Drug Discovery
If you want any further information I’d be happy to try and help.
The origin of the pharmacophore concept.
Any medicinal chemist will use the term “pharmacophore” to describe key features of a ligand binding interaction in 3D, but have you ever wondered where this important concept originated? Thanks to some detective work by Osman F. Güner and J. Phillip Bowen we now have a better idea who the concept originated and evolved. It is all described in a paper in J Chem. Inf. Model DOI.
The IUPAC defines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response”.
It is important to recognise that whilst a specific group of atoms may be used to define a pharmacophoric feature, the steric and electronic requirements can be mimicked by a completely different group of atoms.
Kinase Inhibitors
I’ve updated the Drug Discovery Resources to include a page on Kinase Inhibitors. I will be expanding it over the next week, so any comments or suggestions welcome.
Centre for Therapeutic Target Validation
Target validation is the most critical step in drug discovery because as the chemists will tell you “Most of the other things we can fix”, so I was delighted to hear about the new Centre for Therapeutic Target Validation.
You can read more about it in the Press release
”The Centre for Therapeutic Target Validation is a transformative collaboration to improve the process of discovering new medicines,” says Dr Birney. “The pre-competitive nature of the centre is critical: the collaboration of EMBL-EBI and the Sanger Institute with GSK allows us to make the most of commercial R&D practice, but the data and information will be available to everyone. It is truly exciting to apply so many different areas of expertise, from data integration to genomics, to the challenge of creating better medicines.”
I wish them every success and will be following their work closely.
Drug Discovery Resources Update
I’m in the process of updating the drug discovery resources pages.
I’ve added a couple more examples to the bioisosteres pages and revamped the Computational Chemistry Tools page.
I’ve also updated the Journal RSS feeds and included the feed for Chemical Biology and Drug Design.
Open Source Drug Discovery
If you have ever wondered how Open Source Drug Discovery might work there is a very nice example on the Intermolecular blog here.
Well worth a read.
Published Fragment hits
Whilst there are a variety of techniques to measure the properties or diversity of fragment libraries it is interesting to look at the profiles of compounds that actually appear as hits in fragment-based screening campaigns. I’ve been compiling a database of compounds that have been reported as hits in the literature, this database now has >500 entries culled from 150 publications directed at nearly 100 different molecular targets using 18 different detection technologies and might be expected to give some insight into the type of compounds that appear as hits.
Suggested Books
I’ve just updated the list of suggested books.
Included books on bioisosteres and fragment-based screening.
17th RSC/SCI Medicinal Chemistry Symposium
The registrations are coming in for the forthcoming 17th RSC/SCI Medicinal Chemistry Symposium to be held in Cambridge UK (8-11 Sept 2013). Book early to avoid disappointment.

Full details of the scientific programme are available here together with the registration form.
SMARTCyp 2.4 released
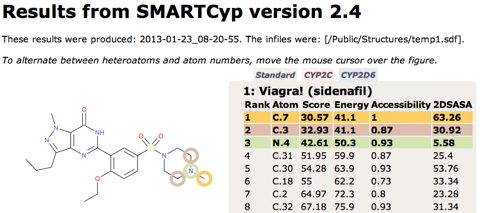
Building a Screening Collection
I’ve just updated the page on building a screening collection and added links to recent useful publications.
Broad Coverage of Commercially Available Lead-like Screening Space with Fewer than 350,000 Compounds, Jonathan Baell DOI
Metal Impurities Cause False Positives in High-Throughput Screening Campaigns, Johannes C. Hermann et al DOI
Drug Discovery Resources Update
I’ve updated the Drug Discovery Resources Pages over the Christmas Break. In particular I’ve updated the Fragment Based Screening section and added a page on building a fragment collection. I’ve also updated the section on CYP interactions, expanding the Induction section.
SMARTCyp Updated
SMARTCyp 2.3 has been released with some additional improvements including: Improved energies for N-oxidations Empirical correction for unlikely N-oxidations of tertiary alkylamines A filtering functionality for excluding compounds with very low activation barriers to CYP-mediated oxidations A smiles string can now be input directly on the command line using the -smiles flag. Available as usual at http://www.farma.ku.dk/smartcyp The science behind the improved N-oxidations and the empirical correction has also been published in a paper in Angewandte Chemie: DOI
Modelling the Drug Discovery Pipeline
An interesting publication in the latest issue of Journal of Cheminformatics in which Melvin Yu uses Monte Carlo simulations to look at the Drug Discovery pipeline DOI. With any analysis of this kind it is easy to argue that it is too simplistic however it does raise some useful discussion points. In particular, the idea that simply attempting to improve drug discovery productivity by simply increasing the size of existing working groups may not necessarily be the best solution.
This also sounds familiar
Simulations also predict that the frequency of compounds to successfully pass the candidate selection milestone as a function of time will be irregular, with projects entering preclinical development in clusters marked by periods of low apparent productivity
Perhaps a greater number of independent smaller research units is a more attractive model?
FiercePharma
FiercePharma are obviously on a recruitment drive, I have to confess I’m already a subscriber. Makes an interesting read over the early morning cup of coffee.
Viewing docking results in Vortex
This may be of interest.
I recently wrote a review of ForgeV10 from Cresset in which I actually imported the results into Vortex to do the analysis. There were however two issues with doing this, firstly interpretation of the 3D structures is sometimes difficult, this can be resolved by creating a 2D rendering of the structure. The other issue is trying to interpret the docking pose whilst looking at the analysis of the results in say a Vortex scatter plot.
I’ve been working with Mike Hartshorn and the people at Dotmatics who have incorporated OpenAstexViewer (a 3D molecule viewer) into the application you can read the full article here..
Drug Discovery Stage Definitions
Whilst the Drug Discovery process is continuous and can vary depending on target it is often useful to split the process into stages with key milestones and targets clearly defined and met before a project moves from one stage to the next.
ODDT Publication
Investigators of rare and neglected diseases can access some of the latest research in the field, plus data about the disorders themselves, through a recently launched free app for Apple devices.
Magic methyl
Methyl Effects on Protein–Ligand Binding
Cheryl S. Leung, Siegfried S. F. Leung, Julian Tirado-Rives, and William L. Jorgensen
A literature analysis of >2000 cases reveals that an activity boost of a factor of 10 or more is found with an 8% frequency, and a 100-fold boost is a 1 in 200 event….The greatest improvements in activity arise from coupling the conformational gain with the burial of the methyl group in a hydrophobic region of the protein.
DOI: 10.1021/jm3003697
ODDT Released
The idea behind ODDT is that there are many rare of neglected diseases that might benefit from collaborative efforts from scientists from multiple disciplines, ODDT is an application that supports informal interactions, provides a means to explore relevant information in a flipboard like interface in particular information tagged by other scientists with similar interests. The image below gives you an idea of the topics currently discussed.
This slideshow explains the genesis of the project, and how it has evolved.
So why not fire up iTunes and download the free app and get involved?
Idiosyncratic toxicity
I’ve updated the section on idiosyncratic toxicity to include more information on risk assessment strategies and the influence of clinical dose.
Fragment Collections
I’ve just completed an addition to the Drug Discovery Resources, since fragment-based screening has increased in popularity I thought I’d have a look at available fragment collections. In particular, I looked at the overlap between collections, the physicochemical profiles and their diversity.
Full details are on the Fragment Collection Profiles page.
ChemSpider Search
If like me you regularly come across drugs mentioned on web pages that you are unfamiliar with then this Safari Extension will be of interest. If a page contains a drug name (this page describes AOX1 substrates), select the name and right click (or control click) and an option appears to search for the highlighted drug on ChemSpider.
Click on Search for “Loratidine” on ChemSpider option and the structure appears in a small window.
The new version of the extension has a few extra options, the small window that pops up containing the structure now has a number of additional options highlighted below, if you click on the “3D” button the display changes to a 3D rendering using the Java applet JMOL. If you now click on the “Zoom” button.
If you then click on the “view” option a new page opens showing the full details in ChemSpider.
Updated Pages
Updated the Chemical Databases and Physicochemical Properties pages of the Drug Discovery Resources.