Building a Fragment Collection
One of the attractions of fragment-based screening is based on the observation “Fragment Space” is smaller than “Chemical Space” and can be more effectively probed with a relatively small library
- A million compounds cover only a small fraction of the suggested 1060 Chemical Space, whilst 2000 compounds can probe much of the 106 Fragment Space
Several approaches have been described in the design of fragment libraries. Most comply with the commonly accepted Astex "Rule-of-Three" (MW <300, H-bond donors/acceptors <=3, cLogP <3). Ideally they should also have solubility measured.
Since fragments would be predicted to have only modest affinity for the molecular target screening has to be carried out at relatively high concentrations. For this reason solubility is an absolutely critical property, whilst there are several algorithms to predict solubility the data generated by Selcia during the construction of their fragment library would suggest they leave something to be desired. The plot below shows calculated solubility (WSKOWWIN software part of the SRC EPI suite) plotted versus the experimental data using a turbidometric assay.
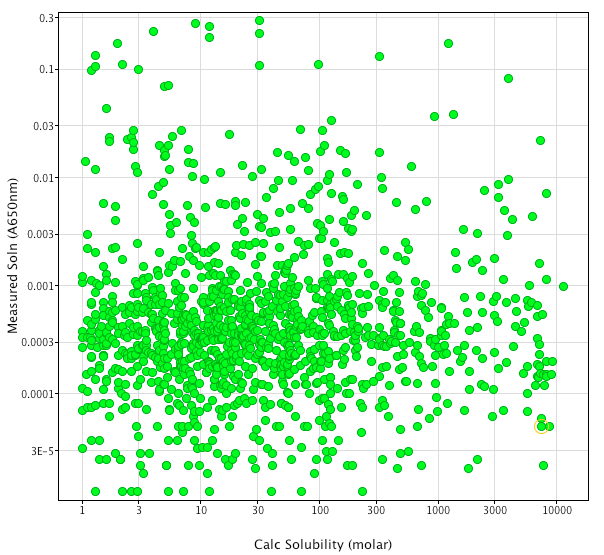
The lack of predictability is perhaps unsurprising since the algorithm was probably trained using larger molecules than these fragments. Selcia and Maybridge have thus taken the approach that all fragments should have measured solubility.
Another consequence of screening at high concentrations is that the influence of impurities can be amplified, careful quality control, particularly the looking for the presence of metals is critical. Strong acids or bases may also overwhelm the buffer solution.
Fragment Library Design
One approach for choosing compounds to be included in the fragment collection is to take a collection of biologically active compounds (e.g. known drugs) and fragment them (e.g. RECAP from CCG), the most common fragments containing 8 or more heavy atoms are then used to search commercial compound collections.
BindingDB is a public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules. BindingDB contains 910,836 binding data, for 6,263 protein targets and 378,980 small molecules.
The first step was to tidy up the structures using sdwash.
sdwash prepares SD files by carrying out a number of operations on the molecular data field, which include 2D depiction layout, hydrogen correction, salt and solvent removal, chirality and bond type normalization, tautomer generation, adjustment and enumeration of protonation states.
The file was then filtered to remove reactive or very high molecular weight compounds.
sdfilter -nonreactive -mw 700- /Users/swain/Documents/Structure_Databases/BindingDB/structures_only.sdf -o /Users/swain/Documents/Structure_Databases/BindingDB/filtered_structures.sdf
Fragmentation using sdfrag, followed by filtering out the very low molecular weight fragments (MWt < 50) resulting in an sdf file containing around 100,000 fragments, many of which were duplicates. This file was then converted to canonical SMILES format using OpenBabel. Datadesk was then used to generate the frequency breakdown for all fragments, the top 100 most common fragments are shown in the table below.
Frequency breakdown of recap
Total Cases 98221 Number of Categories 19721
| Group | Count |
|---|---|
| Cc1ccccc1 | 2802 |
| c1ccncc1 | 2348 |
| C1CNCCN1 | 1815 |
| NC(C)(C)C | 1496 |
| c1ccccc1 | 1351 |
| C1COCCN1 | 1293 |
| c1ccc(cc1)O | 1048 |
| Nc1ccc(cc1)O | 739 |
| CCOC | 729 |
| CC(=O)O | 574 |
| C(F)(F)F | 556 |
| CC(=O)[O-] | 539 |
| Nc1ccccc1 | 526 |
| OCc1ccccc1 | 515 |
| C1CN(CCN1)C | 509 |
| C[S](=O)=O | 505 |
| NC(C=O)C(C)C | 504 |
| c1c(cccc1Cl)Cl | 456 |
| NCc1ccccc1 | 443 |
| N(CC)CC | 432 |
| CCCC | 392 |
| NCCCC | 384 |
| NCC=O | 375 |
| C1CCCN1 | 366 |
| NCC#N | 319 |
| C1CC(CN1)F | 314 |
| OCCOC | 290 |
| C1CCCCN1 | 428 |
| Cc1ccncc1 | 262 |
| Cc1ccc(cc1)O | 255 |
| Cc1ccc(cc1)C#N | 253 |
| c1ccc(cc1)F | 250 |
| Cc1ccccc1O | 250 |
| CCc1ccc(cc1Cl)Cl | 238 |
| Nc1ccc(cc1)Cl | 236 |
| Cc1ccc(cc1)F | 233 |
| C1CCCC1 | 230 |
| C1CCCCC1 | 222 |
| C1NC(CC1Cl)C=O | 222 |
| Oc1ccc2c(c1)CCC2CC(=O)O | 222 |
| c1ccc(s1)Cl | 216 |
| CC(Cc1c[nH]c2c1cccc2)N | 215 |
| NC(C)C | 214 |
| C1CCC(N1)C=O | 212 |
| c1ccc(cc1)OC | 209 |
| Cc1cc(c(cc1)O)O | 199 |
| c1(ccc2c(c1)ccc(c2)Cl)[S](=O)=O | 188 |
| NCCC | 188 |
| CCN(C)C | 184 |
| CCCOC | 183 |
| [S](=O)(=O)c1ccc(cc1)O | 178 |
| Cc1ccc(cc1)Br | 169 |
| Cc1cc(ccc1)C(N)=N | 157 |
| C1CNCCN1CC(=O)O | 156 |
| Nc1ccc(cc1)OC(F)(F)F | 150 |
| CCN1CCOCC1 | 149 |
| C(=O)CC | 148 |
| Cc1ccc(cc1)Cl | 147 |
| C1C(CC(N1)C)F | 144 |
| Oc1ccc(cc1)C(=O)O | 144 |
| C1CNC(C1N)=O | 141 |
| COCCOC | 140 |
| NCC(=O)[O-] | 140 |
| NC1CCNCC1 | 134 |
| Nc1ccc(cn1)Cl | 132 |
| CC1CCCCC1 | 131 |
| C(=O)c1ccccc1 | 130 |
| Nc1cc(ccc1)C | 128 |
| Cc1ccc(cc1)C | 125 |
| Cc1ccccc1OC | 125 |
| C(=O)c1c(cncc1Cl)Cl | 123 |
| CC(C)O | 122 |
| N1C(CC2C(C1)CCCC2)C=O | 122 |
| C1C(NCC1)C#N | 121 |
| CCCO | 121 |
| c1cc(cc(c1)O)O | 120 |
| C1Cc2c(ccc(c2)F)N1 | 117 |
| C(=O)CC#N | 116 |
| Nc1ccc(cc1O)CC=O | 116 |
| Nc1ncccc1N | 116 |
| c1cnc[nH]1 | 113 |
| Cc1c(cccc1F)F | 113 |
| Sc1ccccc1 | 112 |
| [S](=O)(=O)c1ccccc1N | 112 |
| C1NC(CC1Cl)C(=O)NC(C)(C)C | 111 |
Click here if you want to “chemicalize” the page. All SMILES strings will then appear as popup structures.
chemicalize.org is a public web resource developed by ChemAxon which uses ChemAxon's Name to Structure parsing to identify chemical structures on webpages and other text. Related to each structure, structure based predictions are available, as well a search interface is provided to discovery substructures or similar structures
Using the PDB
Whilst the analysis of the BindingDB structures gives us a view of the fragments present in small molecules, we don’t necessarily know much about how these fragments bind to the target protein. So for comparison I also did this with the ligands found as part of the crystal structures in the Protein Data Base http://www.rcsb.org/pdb/home/home.do. In this case we can map the fragments back onto the original ligand(s) and and explore potential binding interactions. The PDB also contains interesting biomolecules that may provide novel fragments. All 125907 identified ligands were downloaded in sdf format, in order to process these ligands it was first necessary to clean up the file using sdwash.
The resulting file was then filtered to screen out ligands containing unusual elements using sdfilter
sdfilter -verbose -expr '"[#T]"=0' -expr '"[!D0!D1!D2!D3!D4]"=0' -elements C,H,N,O,S,P,F,Cl,Br,I /Users/swain/Projects/PDB_ligands/V2000.sdf -o /Users/swain/Projects/PDB_ligands/pass.sdf
The fragments were then generated using sdfrag a that program generates molecular fragments from each molecule in a catenated sequence of SD files. sdfrag is an SVL program intended to be run in MOE/batch. This generated over 4 million fragments many of which were duplicates in a 2.3 GB file.
sdfrag -verbose -ringblock -nounique -recap -schuffenhauer -ringatoms -maxrotbond 4 -simplify /Users/swain/Projects/PDB_ligands/pass.sdf -o /Users/swain/Projects/PDB_ligands/fragments_all.sdf
Handling an sdf file of this size can become an issue so I converted it to canonical SMILES format using OpenBabel, reducing the file size to 32 MB.
/usr/local/bin/babel -isdf '/Users/swain/Projects/PDB_ligands/fragments_all.sdf' -osmi -xc -xn '/Users/swain/Projects/PDB_ligands/fragments_all_struc_only.smi'
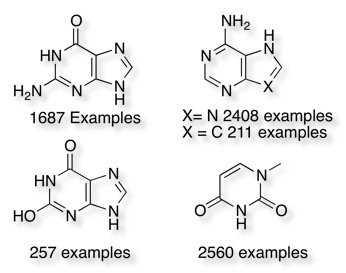
I then opened the file in BBEdit sorted the file and created a separate file of duplicate structures. It then becomes a relatively easy if somewhat tedious exercise to browse through the file looking a series of identical SMILES strings. There were a number of large “fragments” obviously derived from steroids, porphyrins, staurosporin analogues, there were also a large number of amino acids. Perhaps not surprisingly there are also a significant number of nucleotide analogues.
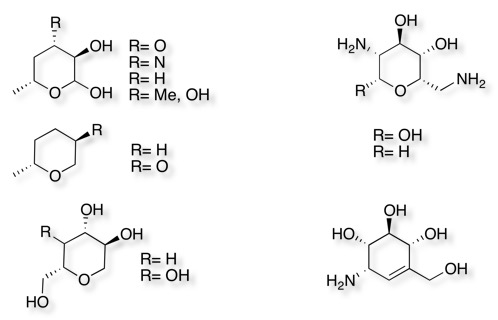
Another group that was very well represented were sugars, with many examples of deoxygenated or amino sugars, these perhaps represent an interesting group of compounds. They are usually very soluble, have a number of groups capable of interacting with the target protein in a usually well defined 3D structure.
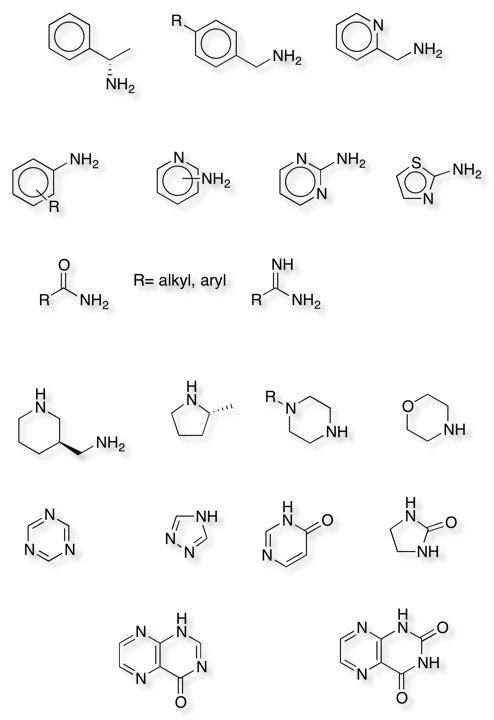
There also a number of organic fragments also appeared regularly, a selection of which are shown below. All contain heteroatoms capable of interacting with the target protein, many contain the sort of functionality that might be expected to interact with a kinase. Notably there are also a significant number of aromatic fragments.
An alternative approach is to take common frameworks or privileged structures (e.g. biphenyl) and decorate them with small functional groups (carboxylate, amine, hydroxyl, halo).
An increasing number of commercial companies are now offering well defined fragments for screening, as might be expected there is significant overlap between the various companies however most also contain unique fragments.
Another approach is to look at fragments that have already been reported as hits.
Updated 5 January 2020