Predicting off-target activities
Small molecules can potentially bind to a variety of bimolecular targets and whilst counter-screening against a wide variety of targets is feasible it can be rather expensive and probably only realistic for when a compound has been identified as of particular interest. For this reason there is considerable interest in building computational models to predict potential interactions. With the advent of large data sets of well annotated biological activity such as ChEMBL and BindingDB this has become possible.
These predictions may aid understanding of molecular mechanisms underlying the molecules bioactivity and predicting potential side effects or cross-reactivity.
Remember 2D similarity methods usually return results in seconds/mins whereas 3D or docking will take hours.
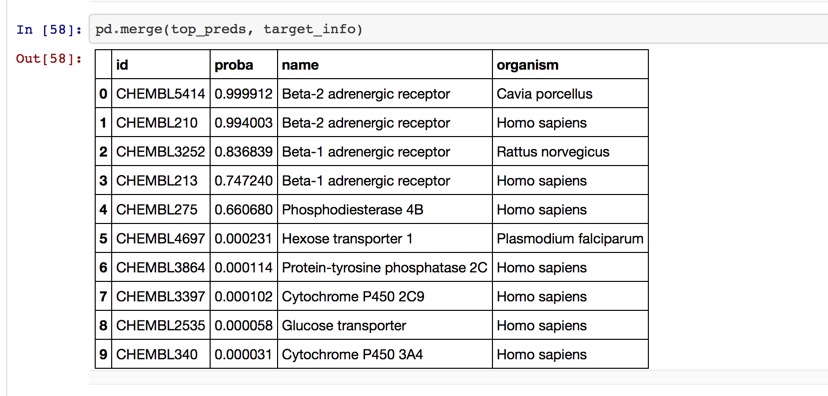
ChEMBL is a great data source for biological activity, the latest update contains 1.6 million unique structures and data on over 11,000 targets taken from 65,213 publications. Not all the target data is suitable for building models however the ChEMBL group have released an Jupyter notebook demonstrating getting target predictions based on the current multi-category Naive Bayesian ChEMBL_21 models for a single molecule input using circular fingerprints. The predictions are ranked by the output classifier probability. Currently there are over 5000 targets modelled.
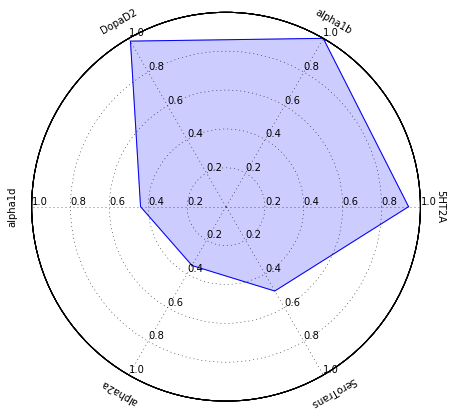
There is also a modified Jupyter Notebook if you are in the situation where you actually want to know the predicted activity at specific targets and display as a radar chart, a graphical method of displaying multivariate data in the form of a two-dimensional chart of multiple quantitative variables each represented on axes starting from the same point.
PIDGIN
Bioactive molecules were extracted from ChEMBL_21 for pChEMBL activity values -Log(Ki/Kd/IC50/EC50) greater than or equal to ‘5’ (10 µm) in binding and functional assays. Percentage activation and inhibition data was also extracted when the ‘Activity Comment’ was declared ‘active’. The dataset contains 766 ,515 active data points, spanning 1,651 human targets. PubChem was mined for inactive compounds in the same procedure to resulting in the extraction of 3 630 485 inactives spanning 1 440 targets
Protein target prediction using Random Forests (RFs) trained on bioactivity data from PubChem (extracted 07/06/18) and ChEMBL (version 24), using the RDKit (2048bit Rdkit Extended Connectivity FingerPrints (ECFP)) and Scikit-learn, which employ a modification of the reliability-density neighbourhood Applicability Domain (AD) analysis by Aniceto DOI. This project is the successor to PIDGIN version 1 DOI and PIDGIN version 2 DOI. 10,446 models generated at four different cut-off’s: 100μM, 10μM, 1μM and 0.1μM for 7,075 biological targets.
Detailed installation instructions are available on GitHub https://pidginv3.readthedocs.io/en/latest/, and can be installed using CONDA
conda create -c keiserlab -c rdkit -c sdaxen --name pidgin3_env python=2.7 pip e3fp scikit-learn=0.19.0 pydot graphviz
A multiple-category Laplacian-modified naïve Bayesian model that was trained on extended-connectivity fingerprints of compounds from 964 target classes in the WOMBAT (World Of Molecular BioAcTivity) chemogenomics database has also been reported DOI. Two algorithms for ligand-target prediction, the Laplacian-modified Bayesian classifier and the Winnow algorithm have been compared using a dataset derived from the WOMBAT database, spanning 20 pharmaceutically relevant activity classes with 13 000 compounds. The supplementary information contains Excel spreadsheets containing all per-class figures of merit for all experiments; comparison of SciTegic’s and our in-house implementation of the Bayesian classifier on a determined partition of the data; Python script used for the calculation of circular fingerprints; the implementations of the Winnow algorithm in C++ and of the Naive Bayesian classifier in Python, as used in the paper. DOI. A novel virtual affinity fingerprinting bioactivity prediction method DPM is based on the docking profiles of ca. 1200 FDA-approved small-molecule drugs against a set of nontarget proteins has been described DOI, the supplementary information contains an example calculation on a small dataset that illustrates the different steps of the DPM method. The increased availability of ligand-target structural information makes the interaction fingerprint an attractive proposition. This method takes advantage of complex structures, either from experimental determination or computational simulation, and extracts qualified atom pairs or atom–residue pairs according to the pre-defined geometric criterion for a variety of nonbonding interactions, such as hydrogen bonds, nonpolar contacts, ionic interactions, π–π stacking, and others.The three-dimensional (3D) structural binding information is then translated into a one-dimensional (1D) binary string, the interaction fingerprint (IF) for the specific ligand–receptor complex DOI.
Many target prediction tools are now available online and the table below highlights a number, giving information on the number of potential targets modelled and an indication of the size and source of the data set. I would recommend avoiding submitting any confidential information to these resources.
| Online Tools | Similarity method | Database | Number Targets | Ref. |
|---|---|---|---|---|
| Polypharmacology Browser | 10 different fingerprints | ChEMBL 21 2.7 million structures | 4613 | [DOI] |
| Polypharmacology Browser2 | nearest neighbours combined with machine learning | ChEMBL 22 2.7 million structures | 1720 | [DOI] |
| PASS Online | Multilevel neighbourhoods of atoms (MNA) descriptors | WDI and ACD | 3678 | [DOI] |
| TarFisDock | Docking | PDTD | 841 | [DOI] |
| Similarity ensemble approach | ECfp4 | CHEMBL 16, WOMBAT, MDDR and StarLite 65,000 structures | Ref. | [DOI] |
| PharmMapper | Receptor-based pharmacophore models | TargetBank, DrugBank, BindingDB, PDTD | 7000 | [DOI] |
| DRAR-CPI | Docking | PDB, DrugBank | 385 | [DOI] |
| TargetHunter | ECfp6, ECfp4 and Openbabel FP2 | ChEMBL 22 and PubChem bioassay 117,535 structures | 794 | [DOI] |
| ChemMapper | Openbabel FP2, MACSS, SHAFT and USR, 2D and 3D similarity | ChEMBL 14, BindingDB, DrugBank, KEGG and PDB 350,000 structures | N/A | [DOI] |
| HitPick | Circular fingerprint FCFP | STITCH 99,572 structures | 1375 | [DOI] |
| SPIDER | CATS and MOE physiochemical descriptors | COBRA 12,661 structures | 251 | [DOI] |
| SwissTargetPrediction | Openbabel FP2 and Electroshape descriptors, 280,000 structures | ChEMBL 16 | 2686 | [DOI] |
| SuperPred | ECfp4, 2D and 3D similarity | ChEMBL, SuperTarget and BindingDB, 341,000 structures | 1800 | [DOI] |
| TarPred | ECfp4 | BindingDB, 179,807 structures | 533 | [DOI] |
| ChemProt | Sfp | ChEMBL 19, BindingDB, DrugBank, PharmGKB, PubChem bioassay, WOMBAT, IUPHAR, CTD and STITCH 1.7 million structures | 2140 | [DOI] |
There is a review of several bioactivity prediction tools here.
Worth reading, Tools for in silico target fishing Methods Volume 71, 1 January 2015, Pages 98–103 DOI.
Last Updated 23 January 2019