
Drug versus Metabolite similarity
A recent paper from Douglas Kell et al DOI has provoked much discussion, especially since it was highlighted on In the Pipeline. The authors suggest that similarity to a human metabolite may be a useful as an indication of how “drug like” a molecule might be.
We exploit the recent availability of a community reconstruction of the human metabolic network (‘Recon2’) to study how close in structural terms are marketed drugs to the nearest known metabolite(s) that Recon2 contains. While other encodings using different kinds of chemical fingerprints give greater differences, we find using the 166 Public MDL Molecular Access (MACCS) keys that 90 % of marketed drugs have a Tanimoto similarity of more than 0.5 to the (structurally) ‘nearest’ human metabolite. This suggests a ‘rule of 0.5’ mnemonic for assessing the metabolite-like properties that characterise successful, marketed drugs. Multiobjective clustering leads to a similar conclusion, while artificial (synthetic) structures are seen to be less human-metabolite-like. This ‘rule of 0.5’ may have considerable predictive value in chemical biology and drug discovery, and may represent a powerful filter for decision making processes.
Whilst this represents an interesting observation I was rather concerned about the choice of a Tanimoto coefficient of 0.5, and decided to repeat the analysis.
The recon-2 dataset was downloaded as a Matlab file, this was exported as a plain text file and Rajarshi Guha converted them to SMILES strings and removed duplicates (and did a comparison with PAINS). I imported these structures into a MOE database and then used a SVL script to compare the recon2 with several other datasets. This included DrugBank that includes details of just under 7000 drug entries, a cleaned up subset of leadlike molecules from Zinc, and BindingDB a public, web-accessible database of measured binding affinities I downloaded in 2008. The datasets were first compared to each other using the MACCS fingerprints with a Tanimoto cutoff of 0.5.
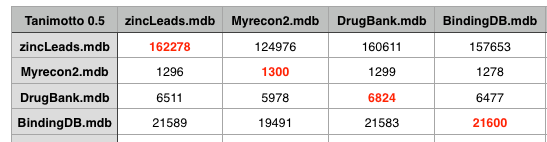
As the table above shows using a Tanimoto coefficient of 0.5 indeed 90% of the molecules in DrugBank are similar to a molecule in recon2, however the same is true for Zinc and BindingDB, indeed at a Tanimoto coefficient of 0.5 all the datasets are pretty similar.
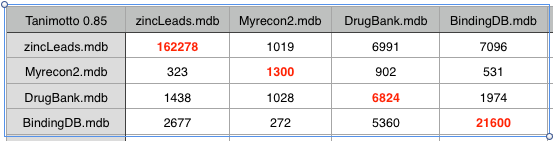
If we increase the Tanimoto coefficient to 0.85 we start to see some resolution, recon2 looks to have more overlap with DrugBank than with either Zinc or BindingDB. However this may simply be a reflection of the fact that DrugBank contains a significant proportion of natural product derived compounds.
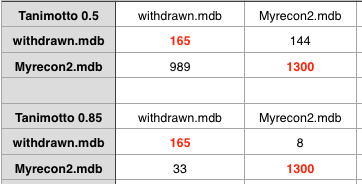
The key question of course is “Does this help us to identify compounds that are likely to fail in development?”. It would be really useful to compare with successful drugs and those that fail in development however I’m not aware of any dataset of of failed drug candidates (if anyone knows of one please let me know). However to in an effort to perhaps get some insight I’ve compared the recon2 set with a dataset of drugs that have been withdrawn (for a variety of reasons). As might be expected using a Tanimoto coefficient of 0.5 offers little discrimination. Increasing to 0.85 it looks like there might be a signal there, but the dataset is too small for firm conclusions.
In summary, this limited exploration suggests there may be something worth following up, but that a Tanimoto of 0.5 simply offers little discrimination.