Kinase Inhibitors (Under Construction)
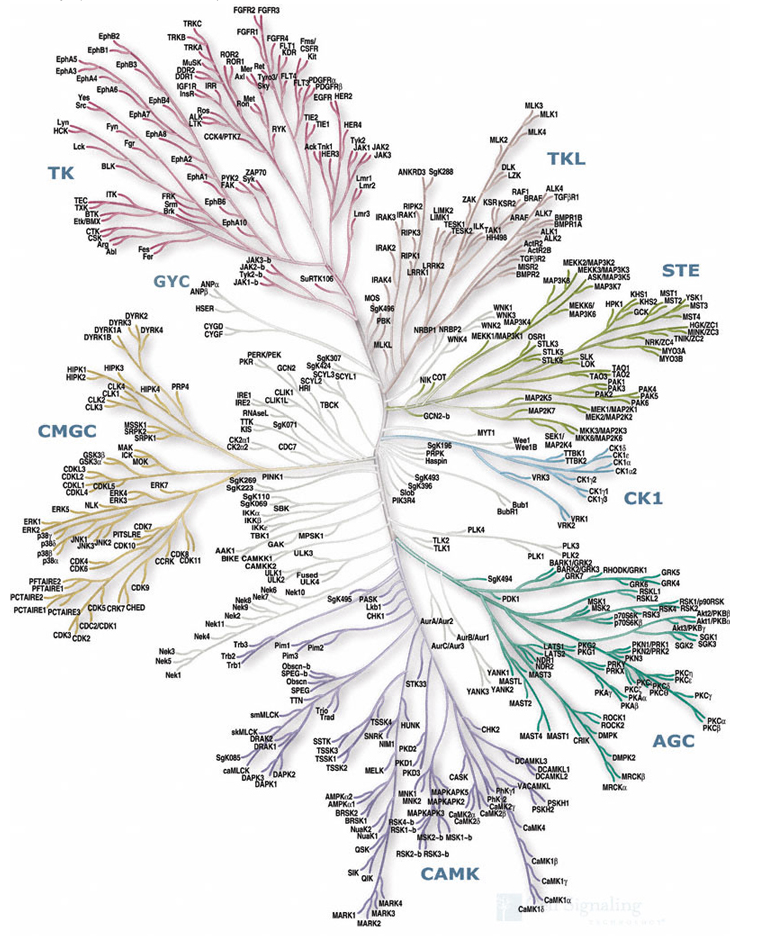
Kinases have become an extremely important molecular target for a range of therapeutic targets. Indeed a search of ClinicalTrials.gov identified 3663 trials with kinase inhibitors. Based on sequence analysis it has been shown that of the 518 human protein kinases, 478 belong to a single superfamily whose catalytic domains are related. The Mannings Group have shown that these can be clustered into groups, families and sub-families, of increasing sequence similarity and biochemical function (Science, 298, 1912-1934) to produce the dendrogram shown below.
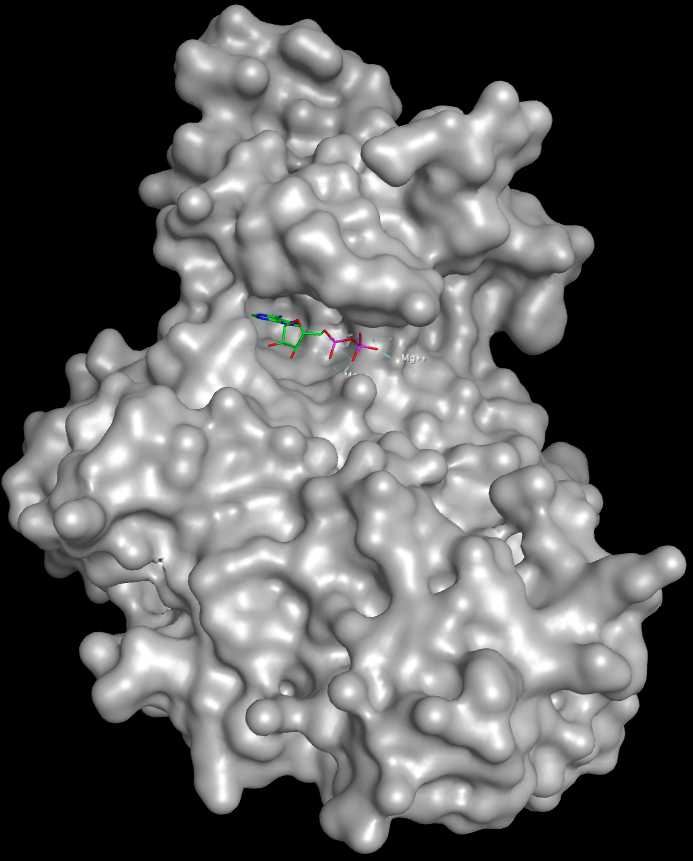
Kinase Binding Site
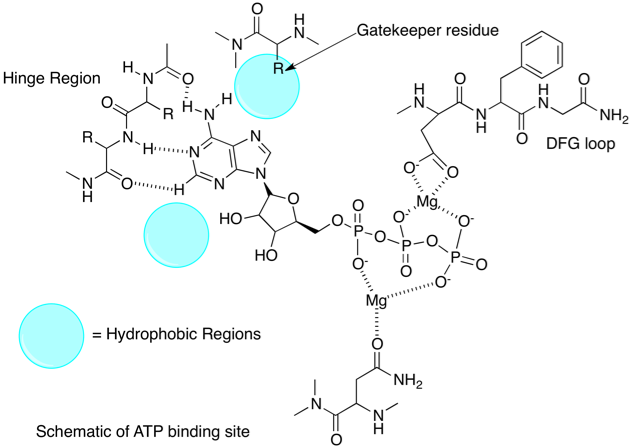
Much of our understanding of the binding of ligands to kinases has come from crystallographic studies, and there are currently several thousand kinase crystal structures in the PDB many for the same protein with different ligands bound. Protein kinases are enzymes involved in the transfer of the terminal phosphate of ATP to substrates that usually contain a serine, threonine or tyrosine residue. Kinases share a conserved arrangement of secondary structure elements that fold into a characteristic twin-lobed catalytic core structure with ATP binding in a deep cleft located between the lobes.
The heteroaromatic adenine ring of ATP then forms a series of hydrogen bonds with the hinge region (shown in yellow below) between the two lobes of the protein.
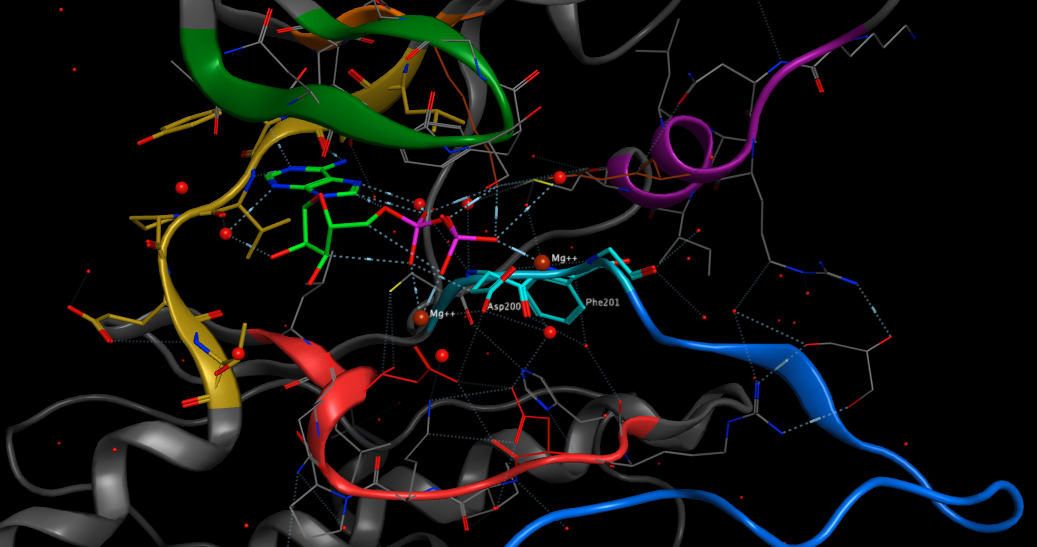
All kinases have a conserved activation loop, which is important in regulating kinase activity and is marked by conserved DFG and APE motifs. At the N terminus of the activation loop there is the conserved DFG motif (shown in pale blue above), also called the magnesium positioning loop because the aspartate (Asp200 above) coordinates magnesium at the active site. This region has been found to be a common regulatory motif in protein kinases. The DFG phenylalanine packs into a hydrophobic pocket between one residue from the N-lobe and one residue from the C-lobe, thus creating a hydrophobic regulatory spine. This packing interaction is referred to as the “DFG-in” conformation. In many kinases, inactivation occurs when this phenylalanine moves out of the hydrophobic pocket, disrupting the orientation of the DFG aspartate and in some cases sterically blocking the ATP binding site . These are referred to as “DFG-out” conformations. The Asp of the DFG is no longer able to coordinate magnesium. The activation loop is shown in dark blue. This is shown schematically below.
Kinase Inhibitors binding
Most of the inhibitors bind in the region of the ATP binding site, probably due to fact that most were discovered using enzyme assays. The Type 1 inhibitors bind to the so called “Active Conformation” of the enzyme and are associated with the DFG-in conformation of this loop. In contrast the Type 2 inhibitors bind to the “Inactive Conformation of the protein, associated with a DFG-out conformation.
Both types of inhibition can be observed for a single protein, and we can compare the binding sites (below). In the image below I’ve overlaid two crystal structures of human LCK P06239, the first (PDB 2ZM1) with the ligand KSF513 shown in free stick display, and the second (PDB 2PLO) with the ligand ST1200 (imatinib) shown in a green ball and stick representation. In the first structure the DFG region and activation loop is highlighted in light blue, it is clear that the phenylalanine of the DFG region occupies volume required for imatinib binding. In the crystal structure of the protein with imatinib bound the DFG region and activation loop is highlighted in dark blue and there is now space for the ligand to bind.
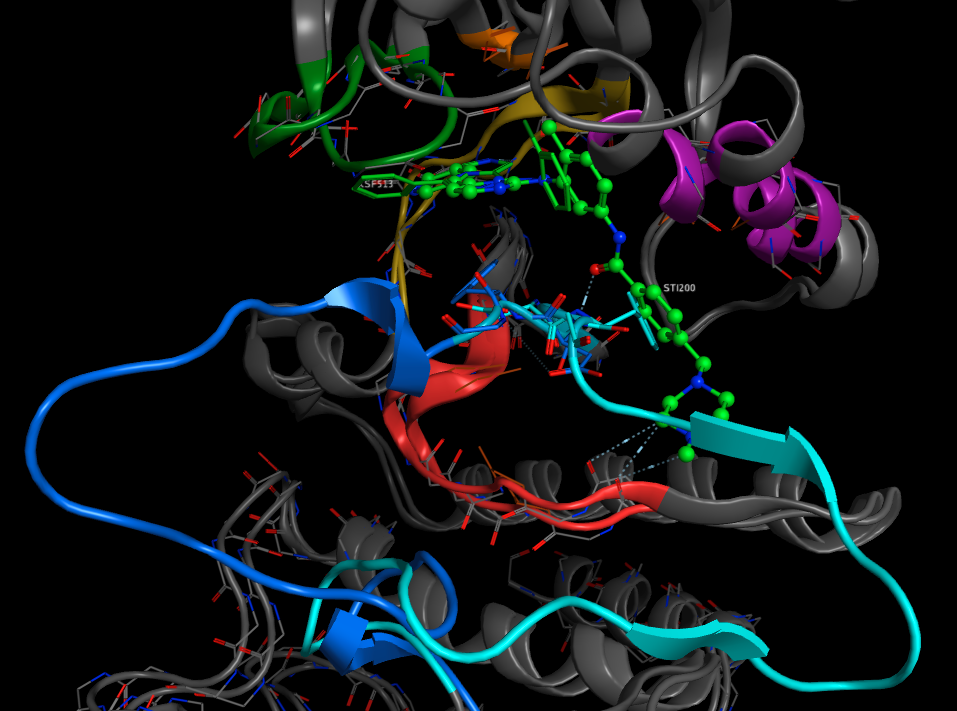
Imatinib is marketed under the name Gleevec and is used for the treatment of multiple cancers including chronic myelogenous leukemia (CML), imatinib works by inhibiting BCR-Abl a protein that is coded for by a fusion gene resulting from a reciprocal translocation between chromosome 9 and 22.
The Gatekeeper
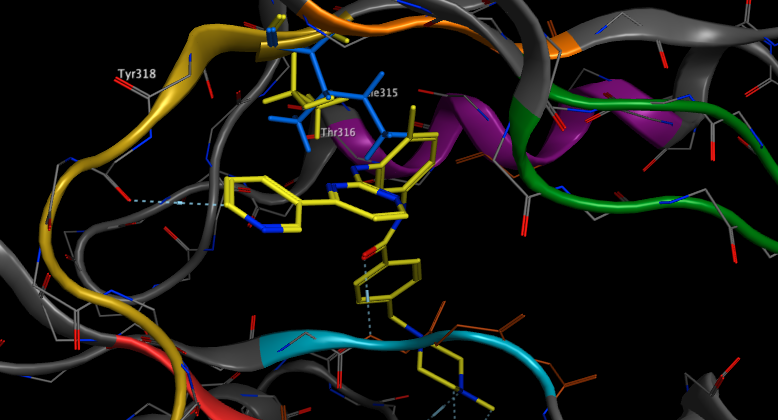
Despite the initial success of Gleevec it was observed that some patients subsequently developed clinical resistance. The mechanism of this resistance was elucidated and shown to be due to previously unknown mutations, in particular a mutation that changes a conserved threonine in the recess of the ATP-binding pocket to another amino acid. Whilst the mutations do not appear to effect catalytic activity they have reduced affinity for inhibitors. The so called ‘gatekeeper’ residue appears to play a role in influencing the sensitivity of inhibitors in numerous protein kinases. In the case of Abl Thr315 (highlighted in yellow below) is mutated to isoleucine (coloured blue below) DOI. It is clear that the increased size of Ile315 prevents imatinib binding.
Allosteric Inhibitors
All the ATP binding site inhibitors have potential issues with selectivity, in some cases there may be an advantage in the inhibition of several different kinases in a signalling cascade in that it reduces the likelihood that a mutation might compromise therapeutic efficacy. In the search for more selective agents scientists have identified allosteric inhibitors, these inhibitors bind to sites remote from the ATP binding site that are unique to a particular kinase.
An interesting example of allosteric binding is seen in the crystal structure of PD 318088 bound to MEK1 1S9J. MEK1 and MEK2 are closely related, tyrosine/threonine protein kinases found in the Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) signalling pathway that has been implicated in a wide variety of cancers.
As the authors highlight DOI
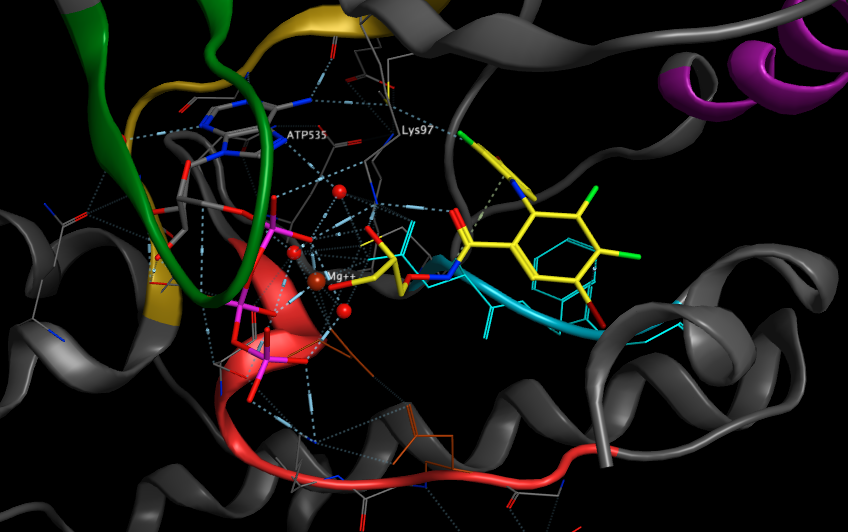
The structures reveal that MEK1 and MEK2 each have a unique inhibitor-binding pocket adjacent to the MgATP-binding site. The presence of the potent inhibitor induces several conformational changes in the unphosphorylated MEK1 and MEK2 enzymes that lock them into a closed but catalytically inactive species. Thus, the structures reported here reveal a novel, noncompetitive mechanism for protein kinase inhibition.
As shown below the inhibitor (shown in yellow) binds in the presence of the ATP-Mg, close to the DFG loop (shown in light blue), interacting with the magnesium and network of waters bound to the triphosphate of ATP.
Covalent inhibitors
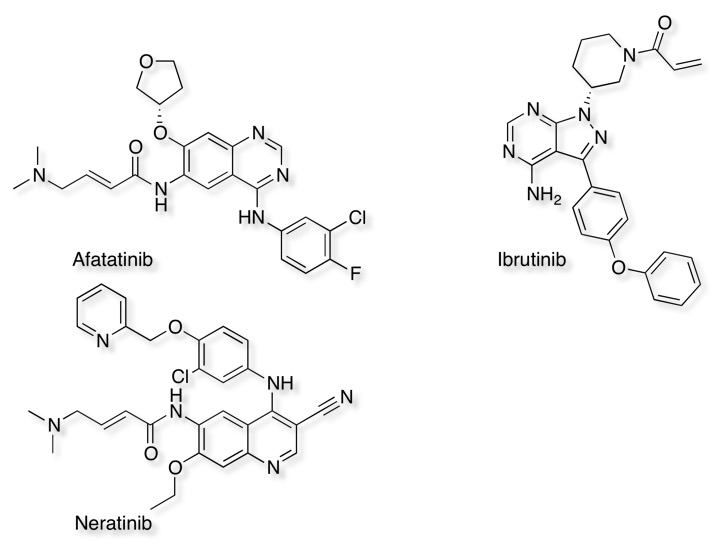
One issue for all ATP-site inhibitors is the need to overcome the high levels of endogenous ATP, whilst allosteric or non-competitive inhibitors offer a solution an alternative is to use "irreversible" inhibitors that are covalently linked to the kinase. The most common mechanism is the Michael reaction, that involves the addition of a nucleophile, such as cysteine, to an α,β-unsaturated carbonyl. The other advantage of irreversible inhibitors is that the half life for inhibition is effectively controlled by the turnover of the enzyme, this could be potentially very useful for indications like cancer where complete suppression of the enzymic activity would be advantageous. Whilst there are concerns that covalent binding to proteins could bring concerns about toxicity the latest compounds are relatively weak electrophiles and require tight binding close to the intended nucleophile for reaction. Three examples of irreversible inhibitors are shown below. An examination of the crystal structures in the PDB would suggest there are >100 different kinases that have a cysteine close to the active site.
There is a page on the Drug Discovery Resources that reviews many different types of covalent inhibitors, and the steps that can be taken to mitigate risk.
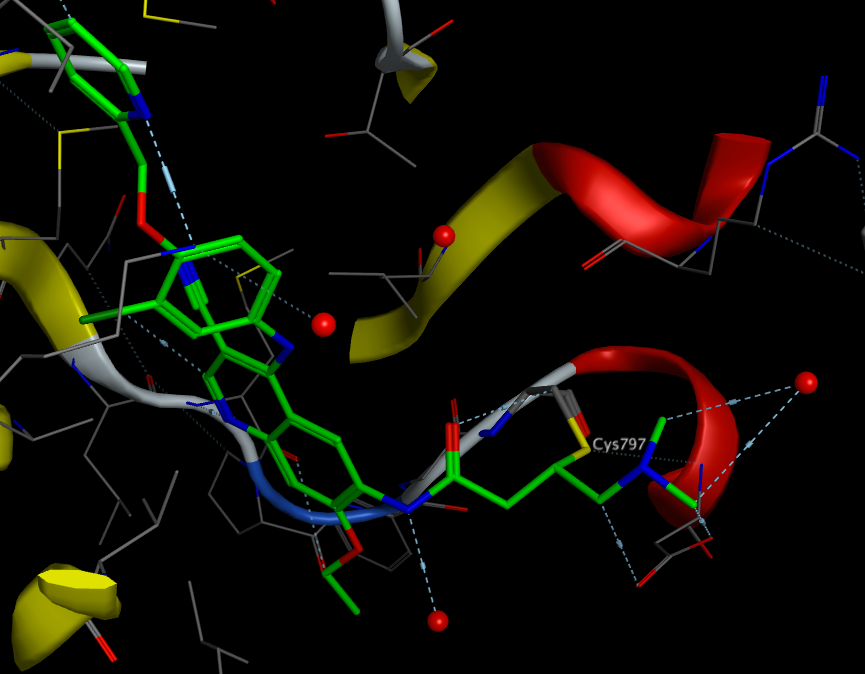
The image below shows Neratinib bound to the active site Cys797 3W2Q
Fragment-based inhibitors
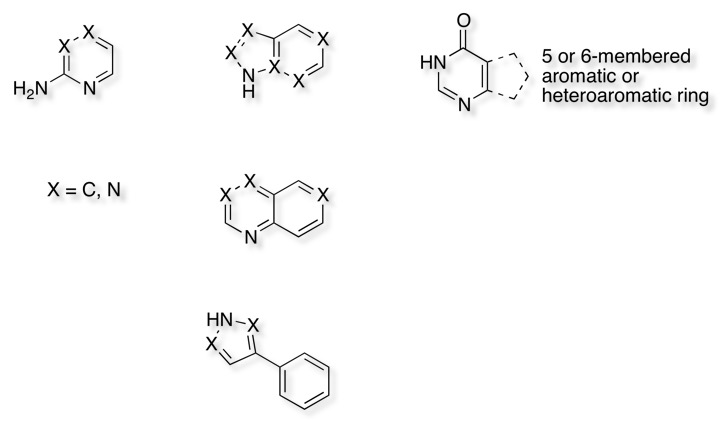
Fragment-based screening has become an increasingly popular means to identify novel starting points for drug discovery programs, indeed 108 of the published fragment hits were identified when screening against kinases. Looking at the structures of the fragments identified it is clear they fall into a limited number of structural classes, the most common are shown in the scheme below. All appear to bind to the ATP binding site, hydrogen bonding to the hinge region.
A recent publication highlights 7-Azaindole: A Versatile Scaffold for Developing Kinase Inhibitors DOI and whilst 7-azaindole does appear as a structural feature in a number of known kinase inhibitors it has also been identified during a fragment screening campaign "Identification of 4-(4-Aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as Selective Inhibitors of Protein Kinase B through Fragment Elaboration" DOI. A number of the known fragment kinase hits are in the PDB and all appear to bind to the hinge region as shown below.
The issue of selectivity
Given that many of the inhibitors target the ATP binding site it is perhaps not surprising that many molecules inhibit multiple kinases, unfortunately this information is not in a readily searchable format. A recent publication "The target landscape of clinical kinase drugs" DOI describes an approach to provide better understanding.
Kinase inhibitors are an important class of drugs that block certain enzymes involved in diseases such as cancer and inflammatory disorders. There are hundreds of kinases within the human body, so knowing the kinase “target” of each drug is essential for developing successful treatment strategies. Sometimes clinical trials can fail because drugs bind more than one target. Yet sometimes off-target effects can be beneficial, and drugs can be repurposed for treatment of additional diseases. Klaeger et al. performed a comprehensive analysis of 243 kinase inhibitors that are either approved for use or in clinical trials. They provide an open-access resource of target summaries that could help researchers develop better drugs, understand how existing drugs work, and design more effective clinical trials.
The results of this analysis are very interesting
To this end, we used a chemical proteomic approach (kinobeads) and quantitative mass spectrometry to characterize the target space of 243 clinical KIs that are approved drugs or have been tested in humans…..The number of targets for a given drug differed substantially. Whereas some compounds showed exquisite selectivity, others targeted more than 100 kinases simultaneously, making it difficult to attribute their biological effects to any particular mode of action. Also of note is that recently developed irreversible KIs can address more kinases than their intended targets epidermal growth factor receptor (EGFR) and Bruton’s tyrosine kinase (BTK).
Useful Resources/Reading
KLIFS is a kinase database that dissects experimental structures of catalytic kinase domains and the way kinase inhibitors interact with them. The KLIFS structural alignment enables the comparison of all structures and ligands to each other. Moreover, the KLIFS residue numbering scheme capturing the catalytic cleft with 85 residues enables the comparison of the interaction patterns of kinase-inhibitors, for example, to identify crucial interactions determining kinase-inhibitor selectivity. DOI.
Kinase SARfari is an integrated chemogenomics workbench focussed on Kinases. The system incorporates and links Kinase sequence, structure, compounds and ChEMBL screening data.
Irreversible kinase inhibitors gain traction Nature Reviews Drug Discovery 12, 649–651 (2013) DOI
All drug profiles can be interactively explored in ProteomicsDB
Updated 4 May 2021