
CYP Interactions
Prediction of Cytochromes P450 Inhibition, Bioinformatics, 2013, 29, 2051-2052 WhichCyp, a tool for prediction of which cytochromes P450 isoforms (among 1A2, 2C9, 2C19, 2D6 and 3A4) a given molecule is likely to inhibit. The models are built from experimental high-throughput data using support vector machines and molecular signatures.
Drug versus Metabolite similarity
A recent paper from Douglas Kell et al DOI has provoked much discussion, especially since it was highlighted on In the Pipeline. The authors suggest that similarity to a human metabolite may be a useful as an indication of how “drug like” a molecule might be.
We exploit the recent availability of a community reconstruction of the human metabolic network (‘Recon2’) to study how close in structural terms are marketed drugs to the nearest known metabolite(s) that Recon2 contains. While other encodings using different kinds of chemical fingerprints give greater differences, we find using the 166 Public MDL Molecular Access (MACCS) keys that 90 % of marketed drugs have a Tanimoto similarity of more than 0.5 to the (structurally) ‘nearest’ human metabolite. This suggests a ‘rule of 0.5’ mnemonic for assessing the metabolite-like properties that characterise successful, marketed drugs. Multiobjective clustering leads to a similar conclusion, while artificial (synthetic) structures are seen to be less human-metabolite-like. This ‘rule of 0.5’ may have considerable predictive value in chemical biology and drug discovery, and may represent a powerful filter for decision making processes.
Whilst this represents an interesting observation I was rather concerned about the choice of a Tanimoto coefficient of 0.5, and decided to repeat the analysis.
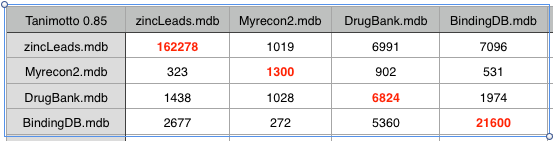
The recon-2 dataset was downloaded as a Matlab file, this was exported as a plain text file and Rajarshi Guha converted them to SMILES strings and removed duplicates (and did a comparison with PAINS). I imported these structures into a MOE database and then used a SVL script to compare the recon2 with several other datasets. This included DrugBank that includes details of just under 7000 drug entries, a cleaned up subset of leadlike molecules from Zinc, and BindingDB a public, web-accessible database of measured binding affinities I downloaded in 2008. The datasets were first compared to each other using the MACCS fingerprints with a Tanimoto cutoff of 0.5.
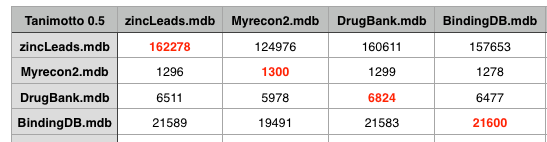
As the table above shows using a Tanimoto coefficient of 0.5 indeed 90% of the molecules in DrugBank are similar to a molecule in recon2, however the same is true for Zinc and BindingDB, indeed at a Tanimoto coefficient of 0.5 all the datasets are pretty similar.
If we increase the Tanimoto coefficient to 0.85 we start to see some resolution, recon2 looks to have more overlap with DrugBank than with either Zinc or BindingDB. However this may simply be a reflection of the fact that DrugBank contains a significant proportion of natural product derived compounds.
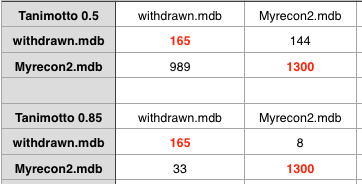
The key question of course is “Does this help us to identify compounds that are likely to fail in development?”. It would be really useful to compare with successful drugs and those that fail in development however I’m not aware of any dataset of of failed drug candidates (if anyone knows of one please let me know). However to in an effort to perhaps get some insight I’ve compared the recon2 set with a dataset of drugs that have been withdrawn (for a variety of reasons). As might be expected using a Tanimoto coefficient of 0.5 offers little discrimination. Increasing to 0.85 it looks like there might be a signal there, but the dataset is too small for firm conclusions.
In summary, this limited exploration suggests there may be something worth following up, but that a Tanimoto of 0.5 simply offers little discrimination.
Bioisosteres pages updated
I’ve updated the pages on bioisosteres to include more examples.
Page on HERG updated
I’ve updated the page on HERG activity, to include a little more information on pharmacophore models.
Fragment sized drugs
As someone who regularly reads Derek Lowe’s “In the Pipeline” blog I was taken with the post on The Smallest Drugs in which he highlighted the structures using the arbitrary cutoffs
the molecular weight cutoff was set, arbitrarily, at aspirin's 180. I excluded the inhaled anaesthetics, only allowing things that are oils or solids in their form of use. As a small-molecule organic chemist, I only allowed organic compounds - lithium and so on are for another category.
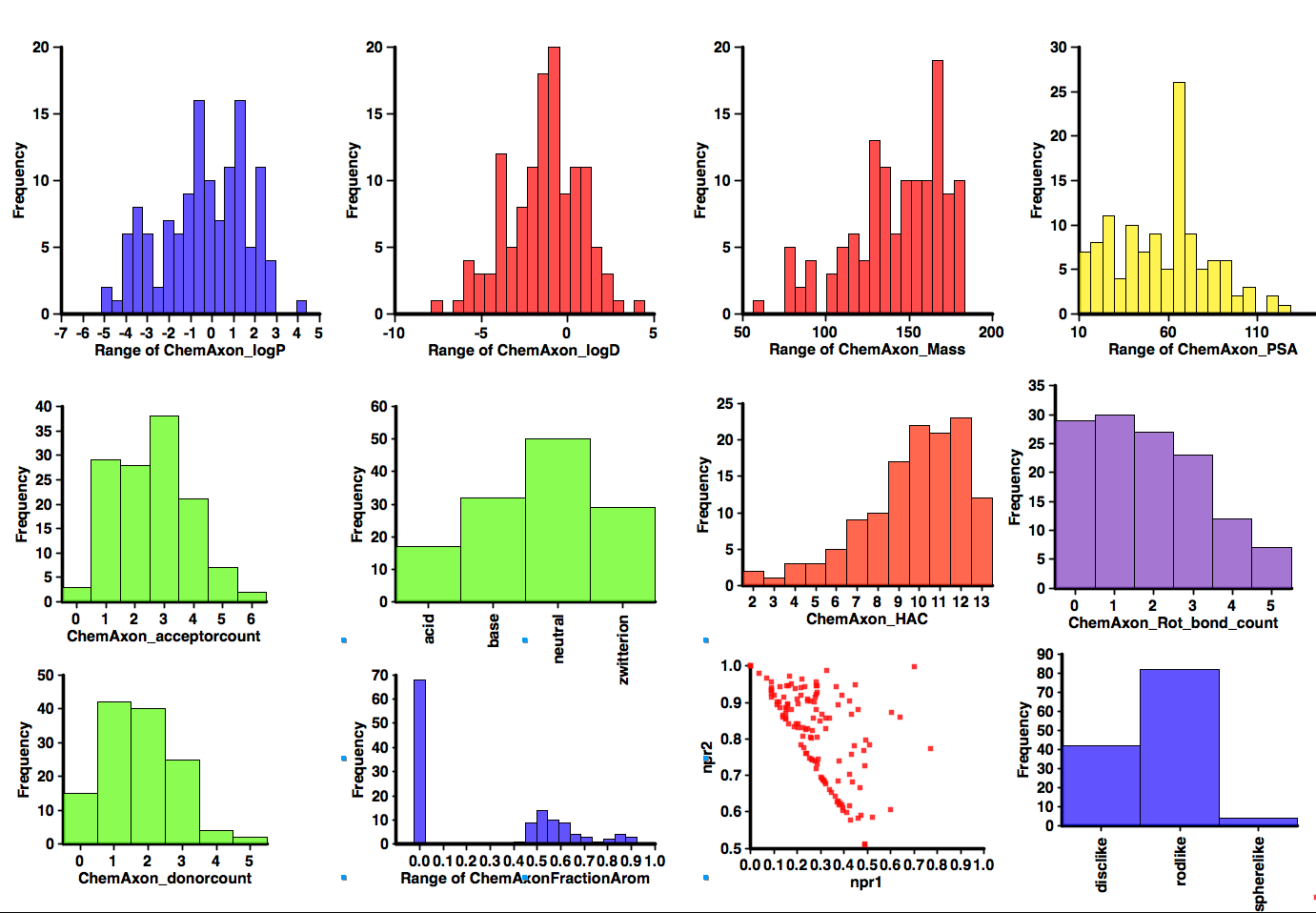
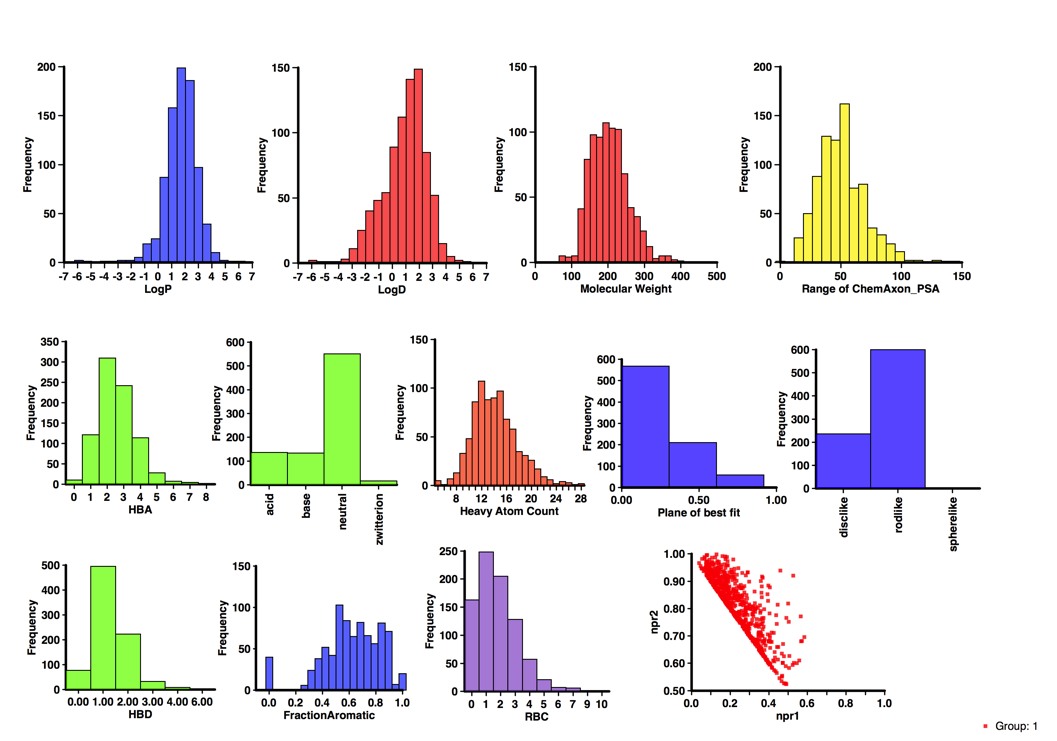
An interesting selection but I thought it might be interesting to profile the calculated properties, I used the DrugBank Database too ensure I got a more comprehensive dataset and then calculated properties as I have done for the Fragment collections. The results are shown below. Probably the most notable feature is the number that contain ionisable groups, over 60% of the molecules would be predicted to be ionised at physiological pH (note however it does include a couple of natural amino acids). Around 50% contain an aromatic ring (of which 2/3 are heterocycles). There are a couple of structures with more 3D shape (Memantine) but in general they would be classified as disc or rod-like. In general the results don’t look too dissimilar to the Published Fragment Hits.
Published Fragments
I’ve updated the page on published fragments, the dataset now includes over 800 published fragments hits abstracted from over 200 publications directed at nearly 130 different molecular targets using 22 different detection technologies and might be expected to give some insight into the type of compounds that appear as hits. With the caveat that the dataset only includes information that has been published.
Seven pharma companies provide access to stalled development compounds
UK researchers will be granted access to a ‘virtual library’ of deprioritised pharmaceutical compounds through a new partnership between the Medical Research Council (MRC) and seven global drug companies, announced today by Business Secretary Vince Cable.
AstraZeneca, GlaxoSmithKline, Janssen Research & Development LLC*, Lilly, Pfizer, Takeda and UCB will each offer up a number of their deprioritised molecules for use in new studies to improve our understanding of a range of diseases. A full list of available compounds will be published later this year, when UK scientists will be able to apply for MRC funding to use them in academic research projects.
This has the potential to a really exciting resource for scientists to explore the pathways involved in a variety of different diseases, and since the compounds have apparently undergone some development it may provide a boon to those involved in repurposing drugs. Much will of course depend on the compounds offered but perhaps other companies will follow suit.
Drug Discovery Resources Updates
I’ve added two new pages to the ADME section, there are now separate pages for CYP2D6 inhibitors and CY3A4 inhibitors.
22 July 2014 Updated to include CYP2C9 and CYP2C19 inhibitors.
18th SCI/RSC Medicinal Chemistry Symposium
18th SCI/RSC Medicinal Chemistry Symposium Sunday 13 - Wednesday 16 September 2015 Churchill College, Cambridge , UK
Europe’s premier biennial Medicinal Chemistry event, focusing on first disclosures and new strategies in medicinal chemistry. Reflecting current trends in medicinal chemistry and pharmaceutical research, the theme of the conference will be ‘Drugging the Undruggable’.
A number of conference places will be reserved for poster presenters and contributions are invited from the whole field of medicinal chemistry. Those presenting a poster may also elect to advertise their poster via oral presentation of a single slide ‘flash’ poster. In addition to traditional plenary talks the organising committee wishes to solicit short talks (20 minutes) describing highly impactful but possibly less complete episodes of medicinal chemistry.
Further Information SCI Conference Dept, 14/15 Belgrave Square, London, SW1X 8PS T: + 44 (0)20 7598 1561 E: conferences@soci.org W: www.soci.org
Longitude Prize 2014
In 1714 the British government threw down the gauntlet to solve the greatest scientific challenge of the century – how to pinpoint a ship’s location at sea by knowing its longitude. Three hundred years later the Longitude Prize 2014 is a challenge with a £10 million prize fund to help solve one of the greatest issues of our time. It is being run and developed by Nesta, with the Technology Strategy Board as launch funding partner.
There are six potential areas highlighted all very worthy causes, however there can only be one prize winner and this is your chance to vote for your preferred project.
The Challenges
WATER How can we ensure everyone can have access to safe and clean water? Water is becoming an increasingly scarce resource. 44 per cent of the world’s population and 28 per cent of the world’s agriculture are in regions of the world where water is scarce. The challenge is to alleviate the growing pressure on the planet’s fresh water by creating a cheap, environmentally sustainable desalination technology.
ANTIBIOTICS How can we prevent the rise of resistance to antibiotics? The development of antibiotics has added an average of 20 years to our life. Yet the rise of antimicrobial resistance is threatening to make them ineffective. This poses a significant future risk as common infections become untreatable. The challenge is to create a cost-effective, accurate, rapid, and easy-to-use test for bacterial infections that will allow health professionals worldwide to administer the right antibiotics at the right time.
DEMENTIA How can we help people with dementia live independently for longer? It is estimated that 135 million people worldwide will have dementia by 2050, which will mean a greater personal and financial cost to society. With no existing cure, there is a need to find ways to support a person’s dignity, physical and emotional wellbeing. The challenge is to develop intelligent, affordable integrated technologies that revolutionise care for people with dementia, enabling them to live independent lives.
FLIGHT How can we fly without damaging the environment? If aircraft carbon emissions continue to rise they could contribute up to 15 per cent of global warming from human activities within 50 years. This needs to be addressed in order to slow down climate change and its detrimental effects on the planet. The challenge is to design and build an aeroplane that is as close to zero-carbon as possible and capable of flying from London to Edinburgh, at comparable speed to today’s aircraft.
FOOD How can we ensure everyone has nutritious, sustainable food? One in eight people worldwide do not get enough food to live a healthy and fulfilled life. With a growing population and limited resources, providing everybody with nutritious, sustainable food is one of the biggest global problems ever faced. The challenge is to invent the next big food innovation, helping to ensure a future where everyone has enough nutritious, affordable and environmentally sustainable food.
PARALYSIS How can we restore movement to those with paralysis? In the UK, a person is paralysed every eight hours. Paralysis can emerge from a number of different injuries, conditions and disorders and the effects can be devastating. Every day can be demanding when mobility, bowel control, sexual function and respiration are lost or impaired. The challenge is to invent a solution that gives paralysed people close to the same freedom of movement that most of us enjoy. Find out more & vote
Drug Discovery Resources Updates
I’ve updated the Drug Discovery Resources, in particular I’ve updated the section on Brain Penetration to include more on predictive models.
I’ve also updated the page on Grant Funding Research.
Worth a look.
The a third edition of the popular book, The Organic Chemistry of Drug Design and Drug Action by Silverman and Holladay has just been released, I’ve added it to the book list.
Vortex users might be interested in a new script that implements an interesting paper from Wagner et al Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties DOI that describes an algorithm to score compounds with respect to CNS penetration.
Lilly MedChem rules can now be installed using Homebrew. In late 2012 Robert Bruns and Ian Watson published a paper entitled Rules for Identifying Potentially Reactive or Promiscuous Compounds DOI. This article describes a set of 275 rules, developed over an 18-year period, used to identify compounds that may interfere with biological assays, allowing their removal from screening sets.
Drug Discovery Resources Updates
I’ve made a couple of updates to the Drug Discovery Resources pages. In particular I’ve updated the Published fragments Hits to include more examples, details of “promiscuous” compounds and summary of detection technologies and the targets explored. I’ve also updated the Aspartic Protease inhibitors page.
As ever comments and/or suggestions very welcome.
Bringing Open Source to Drug Discovery demo
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. I’ve now recorded the demo I showed and put it on YouTube
https://www.youtube.com/watch?v=sG9vDIfp0NE&feature=youtu.be
If you want any further information I’d be happy to try and help.
Bringing Open Source to Drug Discovery
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. As I promised at the meeting I’ve published the slide deck that now includes 25 pages on links and resources that I hope you will find useful.
Bringing Open Source to Drug Discovery
If you want any further information I’d be happy to try and help.
The origin of the pharmacophore concept.
Any medicinal chemist will use the term “pharmacophore” to describe key features of a ligand binding interaction in 3D, but have you ever wondered where this important concept originated? Thanks to some detective work by Osman F. Güner and J. Phillip Bowen we now have a better idea who the concept originated and evolved. It is all described in a paper in J Chem. Inf. Model DOI.
The IUPAC defines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response”.
It is important to recognise that whilst a specific group of atoms may be used to define a pharmacophoric feature, the steric and electronic requirements can be mimicked by a completely different group of atoms.
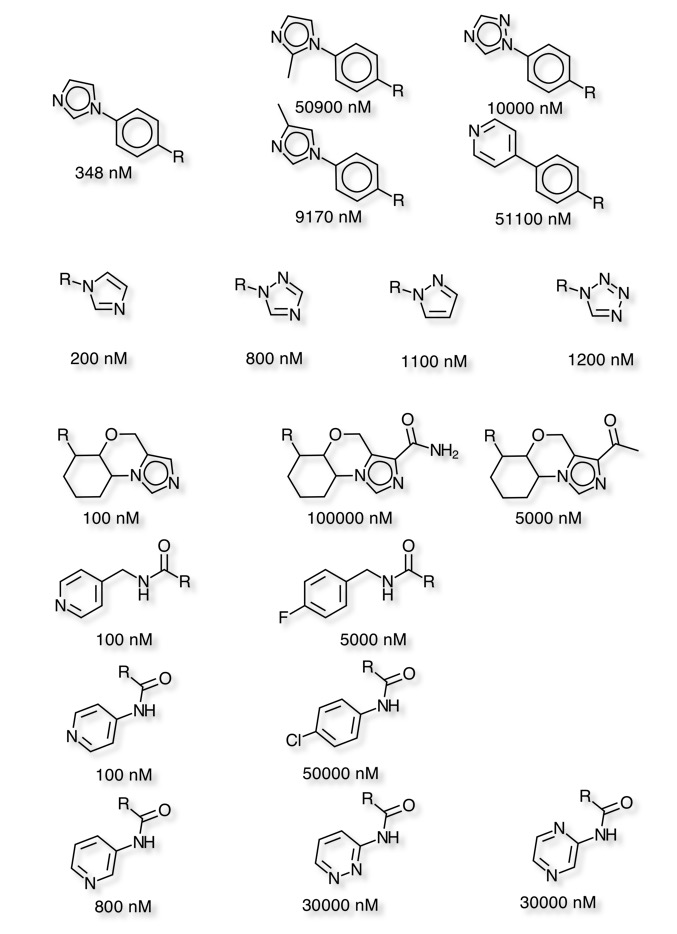
Kinase Inhibitors
I’ve updated the Drug Discovery Resources to include a page on Kinase Inhibitors. I will be expanding it over the next week, so any comments or suggestions welcome.
ChEMBL 18 released
ChEMBL_18 has just been released.
It can be downloaded from the ChEMBL FTP site, and there are more details on the ChEMBL blog
- 1,566,466 compound records
- 1,359,508 compounds (of which 1,352,681 have mol files)
- 12,419,715 activities
- 1,042,374 assays
- 9,414 targets
- 53,298 documents
They now include epigenetic targets, and several new web services giving drug approvals and mechanisms.
Centre for Therapeutic Target Validation
Target validation is the most critical step in drug discovery because as the chemists will tell you “Most of the other things we can fix”, so I was delighted to hear about the new Centre for Therapeutic Target Validation.
You can read more about it in the Press release
”The Centre for Therapeutic Target Validation is a transformative collaboration to improve the process of discovering new medicines,” says Dr Birney. “The pre-competitive nature of the centre is critical: the collaboration of EMBL-EBI and the Sanger Institute with GSK allows us to make the most of commercial R&D practice, but the data and information will be available to everyone. It is truly exciting to apply so many different areas of expertise, from data integration to genomics, to the challenge of creating better medicines.”
I wish them every success and will be following their work closely.
New fragment libraries
It is interesting to see how commercial fragment libraries are starting to evolve, from simple molecular weight cuts of available chemicals to more careful selection based on physicochemical properties. We now see several interesting design strategies being adopted.
Those based on a screening technology such as the LifeChemicals Fluorine-based library to support 19F NMR-based fragment screening, and the Maybridge Bromo-Fragment Collection a collection of over 1500 bromine containing Maybridge fragments constructed as an aid to X-ray based fragment screening.
Other libraries are designed for specific targets
OTAVA offers you new Chelator Fragment Library that comprises 575 compounds in total, Chelators demonstrate binding affinities suitable for FBLD screening and provide a diverse range of molecular platforms from which to develop lead compounds. Also, the propensity for chelators to bind metal ions allows for better prediction of their probable binding position within a protein active site in the absence of experimental structural data of the complex.
Many attractive drug targets contain a free sulfhydryl group in the active site that confounds functional HTS assays due to its facile, non-specific oxidation leading to target inhibition. AnCore have developed a Targeted Covalent Inhibitor fragment library (TCI-Frag™) containing 100+ Rule-of-3 compliant fragments are conjugated with mildly reactive functionalities. The BIONET CNS Fragment Library is a focused library containing 700 Fragments selected for their suitability for Fragment Based Lead Discovery in the areas of CNS drug discovery and Universal target classes.
I’ve updated the Fragment Collections page
Drug Discovery Resources website updated
I’m in the process of updating the Drug Discovery Resources pages, in particular I’ve updated the Grant Funding resources and Fragment screening.
Free online MedChem course
I’ve just been sent details of a new medicinal chemistry course.
Medicinal Chemistry: The Molecular Basis of Drug Discovery
This course explores how to bring a drug from concept to market, and how a drug's chemical structure relates to its biological function. The course opens with an introduction to the drug approval process. This introduction combines the social, economic, and ethical aspects of drug discovery. Topics include how diseases are selected for treatment, the role of animal testing, and the costs of various discovery phases. The course then focuses on the scientific side of drug discovery. Topics include how drugs interact with biological molecules, drug absorption and elimination, and the discovery of weakly active molecules and their optimization into viable drugs.
The course starts 10 March, it is estimated the course will require 6-8 hours per week and runs for 7 weeks. The course was organised by Erland Stevens who wrote the medchem textbook Medicinal Chemistry: The Modern Drug Discovery Process.
Play to Cure™: Genes in Space
Beating cancer through a space game never seemed possible. Until now….
Every day, scientists across the globe are painstakingly analysing the genetic faults in thousands of cancer samples. They are looking for clues that will help develop new cancer treatments. This game let’s you help.
Play to Cure™: Genes in Space is a pioneering way of helping these scientists in their mission to beat cancer sooner and all via this world first mobile game supported by Cancer Research UK.
A mysterious substance is discovered in the voids of deep space. Dubbed Element Alpha, the substance is refined for use in medicine, engineering and construction and soon the Element Alpha industry explodes galaxy wide…..
Aldehyde Oxidase page updated
I’ve updated the page on Aldehyde Oxidase, an enzyme in metabolism of a wide variety of nitrogen heterocycles.
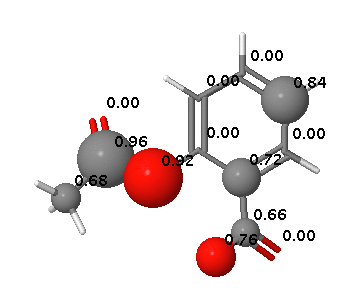
I’ve also included A recent publication DOI that suggests a simple test for the early identification of heteroaromatic drug candidates that have a high probability of metabolism by AO. Bis(((difluoromethyl)sulfinyl)oxy)zinc (DFMS) was used as a source of the CF2H racial, simple LCMS was then used to identify a characteristic M+50 peak. It is also possible to scale up and isolate these metabolically blocked compounds and retest them for improved qualities.
Page on HERG updated
I’ve just updated the page on HERG activity, I’ve included the results of matched pair analysis conducted on the database of berg activity that I have been compiling.
A review of FAst MEtabolizer (FAME)
Whilst much computational work is undertaken to support, library design, virtual screening, hit selection and affinity optimisation the reality is that the most challenging issues to resolve in drug discovery often revolve around absorption, distribution, metabolism and excretion (ADME). Whilst we can measure the levels of parent drug in various medium tracking metabolic fate can often be a considerably more difficult proposition requiring significant resources. For this reason prediction of sites of metabolism has become the subject of current interest.
FAME DOI is a collection of random forest models trained on a comprehensive and highly diverse data set of 20,000 small molecules annotated with their experimentally determined sites of metabolism taken from multiple species (rat, dog and human). In addition dedicated models are available to predict sites of metabolism of phase I and II processes.
FAME offers a high performance prediction of sites of metabolism mediated by a wide variety of mechanisms.
Drug Discovery Resources website annual report
The Drug Discovery Resources section of the website is intended to act as a resource for scientists undertaking drug discovery, it was originally simply a web page version of a course I used to give but has been continuously expanded and updated. Since this takes a far amount of time I like to monitor usage to check that it is being used.
In 2013 the Drug Discovery Resources section was viewed by nearly 24,000 unique visitors (there were 9000 unique visitors in 2012), 27% of which made more than one visit. There were 65,000 page views and on average visitors viewed two pages per visit. There were visitors from 141 different countries with the US and UK being the most common.
The most frequently accessed pages were
Drug Discovery Resources
Plasma Protein Binding and Distribution
Molecular Interactions
Formulation
Fragment Screening
The most popular sections were
ADME
Bioisosteres
Hit Identification
The top search queries were
Plasma Protein Binding
LogD
Bioisosteres
ADME
Aldehyde Oxidase