
3rd In Silico Toxicology Conference
The 3rd In Silico Toxicology Conference, supported by the British Toxicology Society (BTS), the Royal Society of Chemistry (RSC) CICAG Group, Lhasa Ltd., and the Cambridge Alliance on Medicines Safety (CAMS) will take place online on 29 September 2022; attendance is free and open to everyone interested.
Topics will include In Silico Toxicology Consortia, Cell Painting, Gene Expression Data, Biomarkers, Interpreting Neural Networks, Drug-Induced Liver Injury/DILI, Skin Sensitization, Animal Histopathology Data, Species Concordance, In Vivo Pharmacokinetics (PK), Molecular Initiating Events (MIEs), Chemicals, Pharma, Food, Read-Across, ... and beyond (see website for the full programme and registration).
Ultra large Chemical Libraries
In a recent blog post Derek Lowe talked about "Virtual Screening Versus the Numbers" https://www.science.org/content/blog-post/virtual-screening-versus-numbers highlighting some of the issues around ultra large chemical libraries.
It seems quite timely that RSC CICAG is organising a meeting on Ultra Large Chemical libraries 10 August 2022 10:00-17:00, Burlington House, London, United Kingdom.
A decade ago a chemical library of a million compounds was considered large but over the last few years there has been a period of continuous growth in the size of both physical and virtual chemical libraries. As the libraries have grown the conventional search technologies have become unsustainable and new technologies are needed. This meeting will look at the challenges and solutions used to design, create, compare and search these ultra-large chemical libraries.
There are more details and registration here https://www.rsc.org/events/detail/73675/ultra-large-chemical-libraries.
It is now open for abstract submission (oral due by May 1st, posters June 2nd).
Registration fees
Delegate member early £95
Delegate non-member early £115
Delegate member std £120
Delegate non-member std £145
Student member early £65
Student non-member early £85
Student member std £90
Student non-member std £110
Precursor Chemicals list
When helping to enhance screening collections I'm sometimes asked to exclude "prohibited precursor chemicals", these are chemicals that might be used in the manufacture of illegal drugs.
The effective control of chemicals used in the illicit manufacture of narcotic drugs and psychotropic substances is an important tool in combating drug trafficking. These chemicals, known as ‘precursors’, also have legitimate commercial uses as they are legally used in a wide variety of industrial processes and consumer products, such as medicines, flavourings and fragrances.
I'm aware of this list on the UK Government website https://assets.publishing.service.gov.uk/...PRECURSORCHARTDomesticJan2014.pdf, and the listing from INCB http://www.incb.org/documents/PRECURSORS/REDLIST/2020/RedList2020E.pdf, however I'm sure it far from complete.
Does anyone know of a more complete listing? Preferably in a chemically intelligent form
In Silico Toxicology' Network Meeting 2019
A new initiative that should be very useful,
An event (with free registration) to foster the In silico Toxicology Community in the UK and beyond. Scientific contributions are as welcome as those on ongoing work, regulatory aspects, industry perspectives, databases, relevant software, journals etc. in the field. This event is meant to stimulate interactions and discussions, hence speakers are asked to present both about in silico toxicology approaches that are already useful to be applied at the current stage, as well as aspects that don't work that well right now, and where future developments are hence needed.
Venue 26 November 2019, Unilever Lecture Theatre, Department of Chemistry, University of Cambridge (CB2 1EW)
Full details and link for registration here In Silico Toxicology' Network Meeting 2019.
ChEMBL 22 Released
ChEMBL 22 has been released. ChEMBL is a database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data).
This version of the database, prepared on 8th August 2016 contains:
- 2,043,051 compound records
- 1,686,695 compounds (of which 1,678,393 have mol files)
- 14,371,219 activities
- 1,246,132 assays
- 11,224 targets
- 65,213 documents
There is more information in the ChEMBL blog post
Cheminformatics for Drug Design: Data, Models & Tools
Still a few places left at the Cheminformatics for Drug Design: Data, Models & Tools meeting organised by SCI's Fine Chemicals Group and RSC's Chemical Information and Computer Applications Group has been extended.
Imperial War Museum, Duxford, UK Wednesday 12 October 2016
Full details are available here https://www.soci.org/Events/Display-Event?EventCode=FCHEM481
Sounds an excellent meeting and you will have a chance to look around the aircraft at the Duxford Imperial War Museum.
Cheminformatics for Drug Design: Data, Models & Tools
I’ve just heard that the poster deadline for the Cheminformatics for Drug Design: Data, Models & Tools meeting organised by SCI's Fine Chemicals Group and RSC's Chemical Information and Computer Applications Group has been extended.
Imperial War Museum, Duxford, UK Wednesday 12 October 2016
Full details are available here https://www.soci.org/Events/Display-Event?EventCode=FCHEM481
Sounds an excellent meeting and you will have a chance to look around the aircraft at the Duxford Imperial War Museum.
Bringing Open Source to Drug Discovery demo
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. I’ve now recorded the demo I showed and put it on YouTube
https://www.youtube.com/watch?v=sG9vDIfp0NE&feature=youtu.be
If you want any further information I’d be happy to try and help.
Bringing Open Source to Drug Discovery
I spoke at the 25th Symposium on Medicinal Chemistry in Eastern England yesterday and gave a talk/demo on integrating Open Source software into Drug Discovery. As I promised at the meeting I’ve published the slide deck that now includes 25 pages on links and resources that I hope you will find useful.
Bringing Open Source to Drug Discovery
If you want any further information I’d be happy to try and help.
A review of FAst MEtabolizer (FAME)
Whilst much computational work is undertaken to support, library design, virtual screening, hit selection and affinity optimisation the reality is that the most challenging issues to resolve in drug discovery often revolve around absorption, distribution, metabolism and excretion (ADME). Whilst we can measure the levels of parent drug in various medium tracking metabolic fate can often be a considerably more difficult proposition requiring significant resources. For this reason prediction of sites of metabolism has become the subject of current interest.
FAME DOI is a collection of random forest models trained on a comprehensive and highly diverse data set of 20,000 small molecules annotated with their experimentally determined sites of metabolism taken from multiple species (rat, dog and human). In addition dedicated models are available to predict sites of metabolism of phase I and II processes.
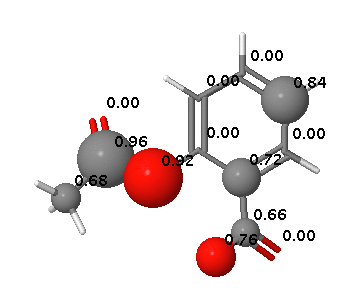
FAME offers a high performance prediction of sites of metabolism mediated by a wide variety of mechanisms.
SMARTCyp 2.4 released
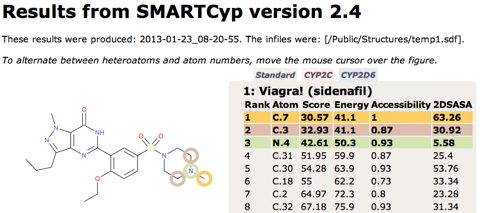
SMARTCyp Updated
SMARTCyp 2.3 has been released with some additional improvements including: Improved energies for N-oxidations Empirical correction for unlikely N-oxidations of tertiary alkylamines A filtering functionality for excluding compounds with very low activation barriers to CYP-mediated oxidations A smiles string can now be input directly on the command line using the -smiles flag. Available as usual at http://www.farma.ku.dk/smartcyp The science behind the improved N-oxidations and the empirical correction has also been published in a paper in Angewandte Chemie: DOI
Viewing docking results in Vortex
This may be of interest.
I recently wrote a review of ForgeV10 from Cresset in which I actually imported the results into Vortex to do the analysis. There were however two issues with doing this, firstly interpretation of the 3D structures is sometimes difficult, this can be resolved by creating a 2D rendering of the structure. The other issue is trying to interpret the docking pose whilst looking at the analysis of the results in say a Vortex scatter plot.
I’ve been working with Mike Hartshorn and the people at Dotmatics who have incorporated OpenAstexViewer (a 3D molecule viewer) into the application you can read the full article here..
OSIRIS Property Explorer
I recently came across a website that may be of interest:
The OSIRIS Property Explorer shown in this page is an integral part of Actelion's (1) inhouse substance registration system. It lets you draw chemical structures and calculates on-the-fly various drug-relevant properties whenever a structure is valid. Prediction results are valued and color coded. Properties with high risks of undesired effects like mutagenicity or a poor intestinal absorption are shown in red. Whereas a green color indicates drug-conform behaviour.