
EU restrictions on Fluorochemicals
In drug discovery the introduction of fluorine into potential drug candidates is an essential part of the medicinal chemists toolbox. Blocking a metabolic hotspot by replacing a Hydrogen by Fluorine or deactivating and aromatic ring by adding a trifluoromethyl are well established strategies for reducing metabolism, increasing half-life and reducing drug load. There are more details on the metabolism page and influence [pi-stacking interactionshttps://www.cambridgemedchemconsulting.com/resources/molecular_interactions.html).
I have a database of all drugs that have been reported to be in phase 3 trials or later (not all will have made it to market) and it is interesting to see how many contain a fluorine atom of some kind. As expected Aryl F and CF3 are the most common
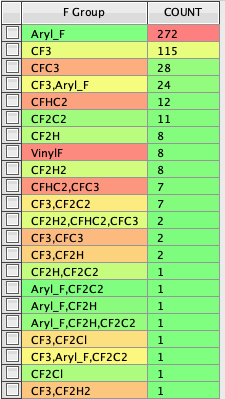
A few examples are shown below.
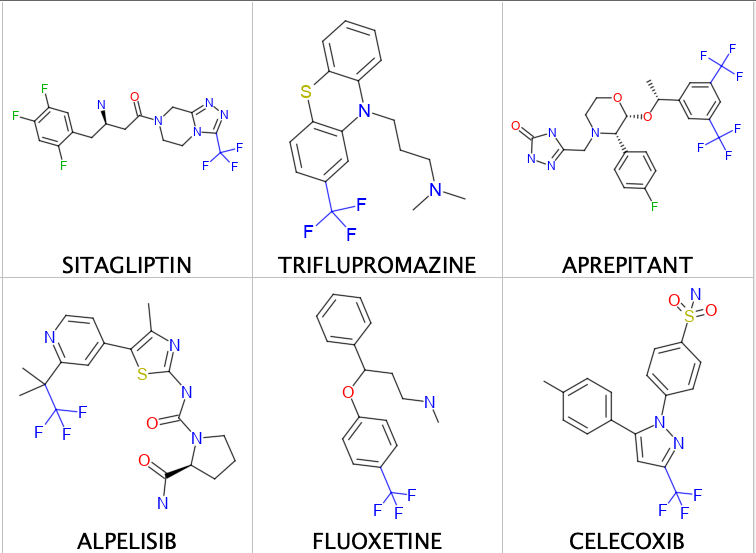
There are also other classes of molecules like perflexane which is used as an imaging contrast agent in echocardiogram. Halothane used clinically as an inhalational anesthetic (on the WHO Model List of Essential Medicines) and other inhaled anesthetics.
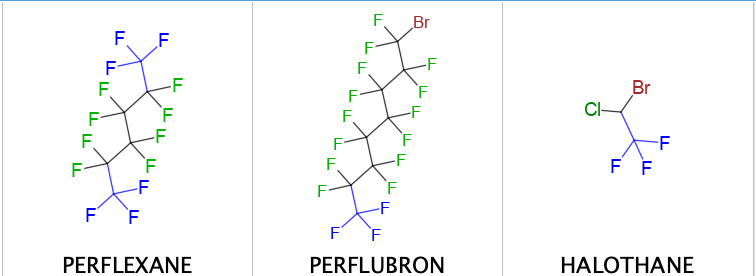
So at first sight the EFMC statement was of some concern.
EFMC STATEMENT ON EU PROPOSAL TO BAN PFAS The EU, through the European Chemicals Agency (ECHA), has launched a proposal that aims to extensively restrict the manufacture, supply, and use of all per- and polyfluoroalkyl substances (Ref 1). PFAS are a large class of synthetic chemicals that are used across a broad range of activities in different scientific areas, not restricted to chemistry. However, several PFAS are environmental pollutants and some of them have detrimental effects on human health. Their extensive use throughout the society, combined with the low reactivity displayed by many fluoroalkyl chemicals, magnifies the potential for accumulation in the environment and contamination of food and water supplies. According to this proposal, which is in public consultation until September 25th (ref 2), PFAS encompasses "any substance that contains at least one fully fluorinated methyl (CF3) or methylene (CF2) without any H, Cl, Br, or I attached to it"
However, looking at the generic scope in more detail it appears that the scope might not be as all encompassing. Looking at the description here https://echa.europa.eu/registry-of-restriction-intentions/-/dislist/details/0b0236e18663449b. The generic scope is described as shown below and I've highlighted a critical phrase.
Per- and polyfluoroalkyl substances (PFASs) defined as: Any substance that contains at least one fully fluorinated methyl (CF3-) or methylene (-CF2-) carbon atom (without any H/Cl/Br/I attached to it).
A substance that only contains the following structural elements is excluded from the scope of the proposed restriction:
CF3-X or X-CF2-X’,
where X = -OR or -NRR’ and X’ = methyl (-CH3), methylene (-CH2-), an aromatic group, a carbonyl group (-C(O)-), -OR’’, -SR’’ or –NR’’R’’’,
and where R/R’/R’’/R’’’ is a hydrogen (-H), methyl (-CH3), methylene (-CH2-), an aromatic group or a carbonyl group (-C(O)-).
My interpretation (caveat I'm not a lawyer) is that the vast majority of drugs, reagents and solvents like trifluoro acetic acid would be excluded. However, inhaled anesthetics and the contrast imaging agents would be included. So the pharma industry needs to be included in the dialog but drugs themselves might have limited impact.
I've not covered polymers, and PTFE is is used in much laboratory equipment from stirrer bars, stopcocks, O-rings, seals etc. and who hasn't used PTFE tape on ground glass joints. There may be alternatives that I'm not aware of, but any substance that has similar properties of inertness, durability etc. may cause the same concerns as PFAs.
The European Chemicals Agency invites interested parties to send in scientific and technical information on the manufacture, placing on the market and use of per- and polyfluoroalkyl substances (PFAS) by 25 September 2023 https://echa.europa.eu/-/echa-seeks-input-on-proposed-pfas-restriction.
Updated Drug Discovery Resources
I have updated several pages in the Drug Discovery Resources.
Ester and Amide Bioisosteres https://www.cambridgemedchemconsulting.com/resources/bioisoteres/ester_bioisosteres.html
Cysteine Protease inhibitors https://www.cambridgemedchemconsulting.com/resources/hitidentification/focus/cysteineprotease_inhibitors.html Covalent Inhibitors https://www.cambridgemedchemconsulting.com/resources/lead_identification/covalent.html.
Bioisosteres pages updated
I've been updating the Drug Discovery Resources. Over the last few days I've been expanding the section on bioisosteres.
A bioisostere is a molecule resulting from the exchange of an atom or of a group of atoms with an alternative, broadly similar, atom or group of atoms. The objective of a bioisosteric replacement is to create a new molecule with similar biological properties to the parent compound. The bioisosteric replacement may be physicochemically or topologically based. The replacement can attenuate toxicity, modify activity of lead, and/or alter pharmacokinetics or the toxicity of the lead.
Bioisosteres are an essential element in the Medicinal Chemists toolbox and the increasing variety reported is a testimony to the creativity of medicinal chemists.
Ester and Amide bioisosteres page updated
A small update to the Ester and Amide bioisosteres page.
Drug Discovery Resources Updated
I've spent a little time updating the Drug Discovery Resources Section of the website. In particular:
- CYP interactions now includes details of published crystal structures and more information on known inhibitors
- I've added page on CYP1A2 from ChEMBL data and updated the CYP2D6 and CYP3A4 pages
- Updated the page on Aldehyde Oxidase
- Added new examples on the bioisosteres page
- Updated the Published Fragments section, including adding the overlay of all examples of Kinase fragment hits from the PDB.
- Added new examples to the Chemical Probes page
- Included more examples of halogen bonding to the Molecular Interactions page
Drug Discovery Resources Updated
I've updated the Bioisosteres section adding a few more examples of aryl ring bioisosteres, and I've added CypReact to the predicting metabolism page.
Bioisosteres database
I just came across a new website that might be interesting, BoBER gives access to a database of bioisosteres.
BoBER (Base of Bioisosterically Exchangeable Replacements) is a freely available method which implements an interface to a database of bioisosteric and scaffold hopping replacements. The web-server enables fast and user-friendly searches for bioisosteric replacements which were obtained by mining the whole Protein Data Bank. BoBER enables medicinal chemists to quickly search and get new and unique ideas about possible bioisosteric or scaffold hoping replacements that could be used to improve specific hit or lead drug-like compounds.
You can submit whole molecule structures which are divided into fragments and then bioisosteric replacements for each fragment can then be identified.
There is a section on bioisosters in the Drug Discovery Resources.
Updated bioisosteres pages
Bioisosteres are an essential tool in the medicinal chemistry toolkit.
Bioisosteres are chemical substituents or groups with similar steric or electrostatic properties which produce broadly similar biological properties to another chemical compound.
Bioisosteres won't always give improved properties, and sometimes we find that a transformation that improves metabolic stability in one series might have the reverse effect in another. However they provide useful (literature precedented) starting points for structural transformations that allow exploration of structure activity relationships. Sometimes they yield similar activity but offer alternative structural vectors for exploration, sometimes they simply improve solubility.
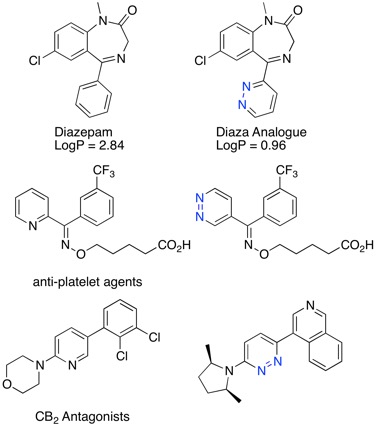
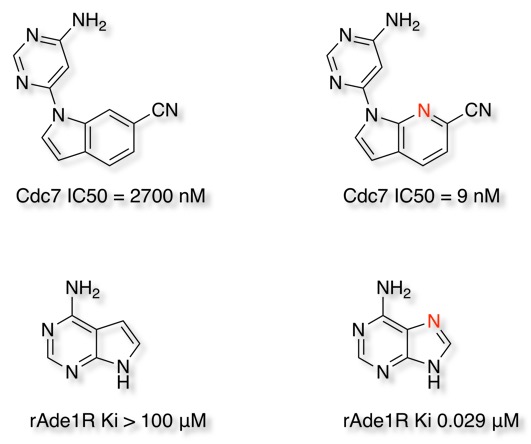
I've now updated the bioisosteres section of the Drug Discovery Resources including new examples people have kindly sent to me.
FDA approves deutetrabenazine
The FDA have approved deutetrabenazine, the deuterated analog of tetrabenazine, for chorea associated with Huntington disease. This represents an example of perhaps the simplest bioisostere and the first example to gain FDA approval.
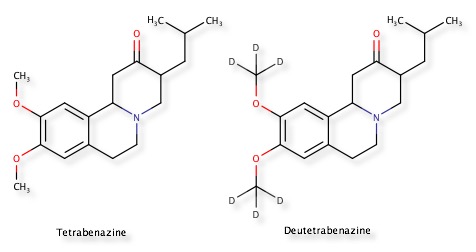
There are several other examples of the H to D change to reduce the rate of metabolism and improve pharmacokinetics.
Bioisosteres Updated
I’ve just updated the aromatic bioisosteres page to include the bicyclo[1.1.1]pentane replacement for phenyl described in a recent publication DOI.
Drug Discovery Resources Update
I’m in the process of updating the drug discovery resources pages.
I’ve added a couple more examples to the bioisosteres pages and revamped the Computational Chemistry Tools page.
I’ve also updated the Journal RSS feeds and included the feed for Chemical Biology and Drug Design.
Nitro bioisosteres.
At the 17th RSC-SCI Medicinal Chemistry Conference in Cambridge Alexander Pasternak (Merck) gave an excellent talk on their work to identify a potent and selective ROM-K inhibitor as novel diuretics. The ROM-K potassium channel is a member of the inward rectifier family of potassium channels expressed in two regions of the kidney: thick ascending loop of Henle and cortical collecting duct DOI, ROMK participates in potassium recycling across the luminal membrane which is critical for the function of the Na+/K+/2C1" co-transporter, the rate- determining step for salt reuptake in this part of the nephron. At the cortical collecting duct , ROMK provides a pathway for potassium secretion that is tightly coupled to sodium uptake through the amiloride sensitive sodium channel. This makes ROM-K an attractive potassium sparing diuretic target.
To cut a long story short Merck ran a HTS campaign (actually I think they ran two) and the only hit is shown below.
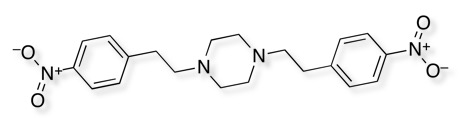
As I am sure all medicinal chemists are aware nitro groups, in particular aromatic nitro groups are well known to be reduced in vivo yielding hydroxylamines and nitrosoamines that are highly reactive species and are known carcinogens. So whilst one nitro in the hit is bad imagine how it feels to have two!
The Merck group however followed this lead up and managed to identify several bioisosteric replacements for the nitro group,
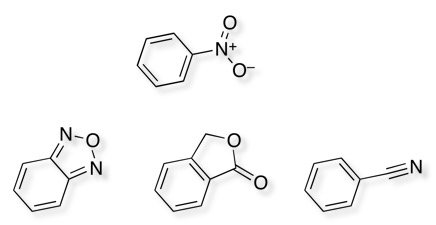
Interestingly there have been two other reported hits for the same target, and these also include nitrobenzenes.
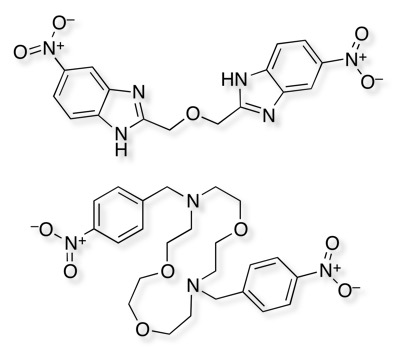
These structures underline the importance of the arylnitro group but also raise a couple of interesting questions, whether nitro compounds should be removed from screening collections? In addition, given the structure of ion channels is often a parallel array of identical proteins forming a pore through the membrane perhaps we should try to populate screening collections with palindromic structures that might bind linking two chains?
I’ll add these to the bioisosteres section at the weekend.
Bioisosteres Pages updated
I’ve added a few more examples to the Bioisosteres pages. Many thanks to the readers who sent the suggestions.
A review of Bioisosteres in Medicinal Chemistry
When I look at the weblogs of the Drug Discovery Resources section of my website it is clear that two sections are of particular interest, Fragment-Based screening attracts a steady stream of readers and the other area that is very popular is the section on Bioisosteres. The former because it is an increasingly popular, effective and low cost entry point into screening for leads, the latter presumably because it provides a great way to generate ideas of what one might do next.
I’m not aware of a book dedicated solely to Bioisosteres so I was delighted to hear about a new book in the area Bioisosteres in Medicinal Chemistry edited by Nathan Brown.
Having had time to read through the book I thought I’d post my thoughts.
A bioisostere is a molecule resulting from the exchange of an atom or of a group of atoms with an alternative, broadly similar, atom or group of atoms. The objective of a bioisosteric replacement is to create a new molecule with similar biological properties to the parent compound, but perhaps with improved physicochemical or ADME properties. Whilst the experienced Medicinal Chemist will have a toolbox of such replacements, this book is an excellent resource that provides a logical framework to this area of drug discovery that will be useful to all.
The book is organised into four section, the first deals with classical replacements, the second the discovery of bioisosteres based on database mining, the third section deals with physicochemical properties and shape, the final section includes the application of bioisosterism in a drug discovery project.
The book starts by giving a short history of bioisosteres and several examples of the attempts to define what is exactly meant by a bioisostere, it then quickly moves onto description of the development of many of the classical bioisosteres, giving plenty of examples and references. The influence of the bioisostere on ADME properties is also introduced again with plenty of key examples. The second section starts by describing the evolution of the Bioster database that has been created by abstracting examples from the Medicinal Chemistry literature. There is also a description of how it might be used as a source of bioisosteric replacements. In contrast mining the Cambridge Structural Database has the potential to identify unexpected replacements based on the arrangement of potential non-bonding interactions. As mentioned agove one of the key drivers for looking at bioisosteres is to improve ADME properties, and mining a large database of metabolic stability data has allowed the identification of transformations that are more likely to improve stability. The third section deals with physicochemical properties, molecular topology and shape, describing in some detail the in silico techniques that can be used to scaffold hop to identify novel frameworks. The final section describes the Drug Guru project, and the use of the molecular field based screening in a NPY-Y5 project.
In summary the book is an excellent starting point for new medicinal chemists and a useful resource for more experienced scientists to dip into to generate ideas. in summary every chemist involved in drug discovery would benefit from access to this book.
Bioisosteres in Medicinal Chemistry
I mentioned this book in the past and I have just been told it is now available on Amazon.
Written with the practicing medicinal chemist in mind, this is the first modern handbook to systematically address the topic of bioisosterism. As such, it provides a ready reference on the principles and methods of bioisosteric replacement as a key tool in preclinical drug development. The first part provides an overview of bioisosterism, classical bioisosteres and typical molecular interactions that need to be considered, while the second part describes a number of molecular databases as sources of bioisosteric identification and rationalization. The third part covers the four key methodologies for bioisostere identification and replacement: physicochemical properties, topology, shape, and overlays of protein–ligand crystal structures. In the final part, several real–world examples of bioisosterism in drug discovery projects are discussed. With its detailed descriptions of databases, methods and real–life case studies, this is tailor–made for busy industrial researchers with little time for reading, while remaining easily accessible to novice drug developers due to its systematic structure and introductory section.
Bioisosteres Updated
Bioisosteres in Medicinal Chemistry by Nathan Brown
More bioisosteres added
I’ve updated the bioisosteres section, adding a number of new examples of bioisosteric replacements.
More Ester and Amide bioisostere examples
I’ve updated Bioisosteres section adding several new examples to the Ester and Amide bioisosteres.
Piperazine bioisosteres
At the 16th SCI/RSC Medicinal Chemistry Symposium held in Churchill College, Cambridge there we a couple of examples of bioisosteric replacements for piperazine. Since piperazines turn up regularly in sample collections I thought it might be useful to add them to the page of basic bioisosteres.
Updated Bioisosteres
Judging by the weblogs the Bioisosteres section has been very popular so I’ve added several more examples that I hope will be useful.