
9th Fragment-based Drug Discovery Meeting slides
I'm just back from the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ another fabulous meeting and always great to hear about the multitude of ways that Fragments are impacting drug discovery, from target identification, hit discovery to lead optimisation. A number of people asked if the slides from my talk would be available.
Hopefully this link will be accessible to everyone.
http://cambridgemedchemconsulting.com/news/files/FragHitsMar2024.pdf.
9th Fragment-based Drug Discovery Meeting
I'll be heading over to the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ later today. This event is one of the high points in the Drug Discovery calendar. I'm sure there will be plenty to add to the Fragment-Based Screening section on the Drug Discovery Resources Website.
The aim of the 9th RSC-BMCS Fragment-based Drug Discovery meeting will be to continue the focus on case studies in Fragment-based Drug Discovery that have delivered compounds to late stage medicinal chemistry, preclinical or clinical programmes. The Fragment series was started in 2007 and continues with this theme in having over three-quarters of the presentations focused on case studies. This will be complemented by technology progress in high concentration, NMR, SPR and X-ray screening.
Binding Sites are 3D
I've always found it interesting that whilst everyone recognises that protein binding sites are three dimensional (and chiral) there is a reluctance to have chiral centres in screening hits. This is despite examples were chiral centres aid affinity, selectivity and solubility. I suspect one of the concerns is the ease of follow up for any hits.
I've been working with Liverpool Chirochem to design a 3D rich, homochiral fragment screening library. The real beauty of this library is that the fragments can be easily expanded using validated chemistry in their parallel synthesis lab.
Once you have built a supply of these homochiral building blocks they can of course be put to many different additional uses, covalent fragments, DEL building blocks, and as building blocks for large virtual libraries. All of which can be supported by their parallel synthesis lab.
An interesting opportunity
Postdoctoral Scientist for Protein Crystallography SRF Centre for Medicines Discovery (CMD), Biochemistry Phase II, South Parks Road, OX1 3QU We are seeking to appoint a Postdoctoral fellow for the Protein Crystallography Small Research Facility (PX-SRF), under the supervision of Professor Frank von Delft.
Do you include chiral fragments in your screen?
Fragment-based screening has become increasingly popular over the last 15 years and has proven to be a viable alternative to high-throughput screening.
A little while ago I posed the question "Do you include chiral fragments in your screen?" And the results were interesting. Whilst many, many folks clicked on the link very few actually responded to the poll, perhaps something people are thinking about but don't have an answer?
For those that did respond it seems that very few actively exclude chiral fragments.
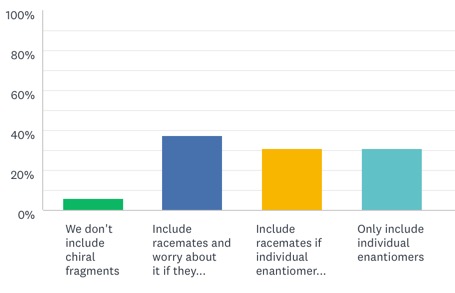
Of those that included chiral fragments the responses were pretty evenly split between those that include racemates and only worry about the enantiomers if they appear as a hit, those that only include racemates if the individual enantiomers are available and those that only include individual enantiomers.
One of the arguments against including chiral fragments has been it increases the complexity of the fragments and as the ligand/receptor match becomes more complex the probability of any given molecule matching falls. However, biological targets are almost invariably chiral and selectivity often relies on stereochemical features.
One of the key attractions of fragment screening is the ability to explore/validate hits without committing significant chemistry resources, so I’d argue that the ready availability of related analogues ought to be part of the library selection criterion and by extension having access to the individual enantiomers should be an important consideration.
Fluorine Fragment Library
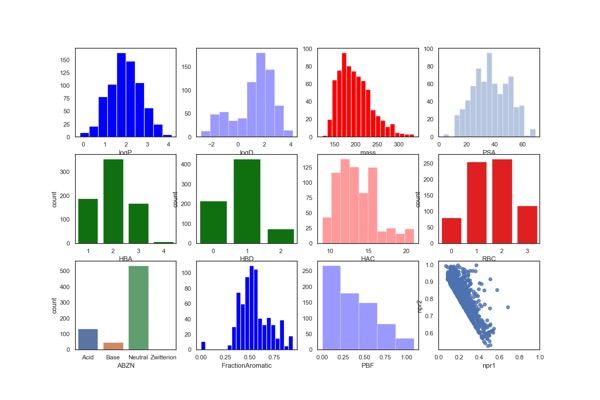
Key Organics have expanded the BIONET Fluorine Fragment Library which now includes 719 fluorinated fragments. All 719 fragments in the Fluorine Fragment Library have been analysed by 19F NMR and 1H NMR for:
- Structure verification
- Purity
- Measured solubility in PBS buffer ≥ 100μM
- Aggregation.
The calculated physicochemical properties of the library are shown below. They have also been filtered using PAINS and Lilly MedChem rules.
There are more Fragment Collections described here.
Chiral Fragment Screening
One of the attractions of fragment-based screening is that “Fragment Space” is smaller than “Chemical Space” and can be more effectively probed with a relatively small library. However, if we include chiral fragments the size of the library increases, as does the complexity of the ligands. It can also add extra work if the individual enantiomers of a chiral fragment are not available.
The poll linked below is intended to try and capture the current view. I'll post the results here later.
Precursor Chemicals list
When helping to enhance screening collections I'm sometimes asked to exclude "prohibited precursor chemicals", these are chemicals that might be used in the manufacture of illegal drugs.
The effective control of chemicals used in the illicit manufacture of narcotic drugs and psychotropic substances is an important tool in combating drug trafficking. These chemicals, known as ‘precursors’, also have legitimate commercial uses as they are legally used in a wide variety of industrial processes and consumer products, such as medicines, flavourings and fragrances.
I'm aware of this list on the UK Government website https://assets.publishing.service.gov.uk/...PRECURSORCHARTDomesticJan2014.pdf, and the listing from INCB http://www.incb.org/documents/PRECURSORS/REDLIST/2020/RedList2020E.pdf, however I'm sure it far from complete.
Does anyone know of a more complete listing? Preferably in a chemically intelligent form
Flatland: a nice place to be
As ever a useful analysis of the published literature on Practical Fragments, "Evaluation of 3-Dimensionality in Approved and Experimental Drug Space" DOI.
The true need for topological diversity in feedstocks and final drug molecules remains unclear given the overwhelming number of linear and planar drugs. The question remains as to whether more 3D compounds represent attractive and untapped therapeutic space, or if more linear/planar molecules are indeed the best topologies for bioactive molecules.
I came to a similar conclusion when looking at published fragment hits.
Fragments and novelty
I've spoken to a couple of people recently who are very focused on identifying novel fragments for their fragment screening collection. I have to say I'm not convinced about the benefit of populating your fragment collection with novel fragments. One of the really attractive features of fragment-based screening is the ability to follow up and verify the initial round of fragment hits by testing commercially available analogues, or isomers.
You can read more here Fragments and novelty.
Fragment hits for SARS-CoV-2
A group of researchers including Dave Stuart, Martin Walsh, and Frank von Delft (Diamond Light Source) has performed a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Even before fully analyzing all of the data they have released interim results https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html.
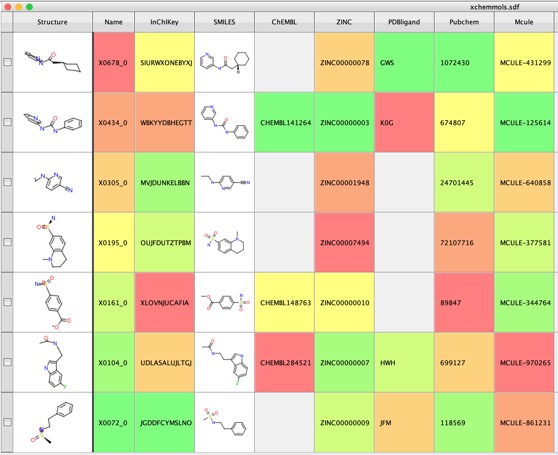
The hits can be viewed on fraglaysis here.
I've downloaded all the structures that were screened, both those that bind and those where no binding was observed and put them into a single file, also added inChiKey, SMILES, PubChem ID, PDB ID of ligand if known and a range of other identifiers from different databases, the file is available here http://cambridgemedchemconsulting.com/news/files/Archive.zip
Fragment based screening pages updated
I spent some time over the Christmas break updating the Drug Discovery Resources pages on Fragment-Based screening, adding new vendors and updating the physicochemical profiles. I've also added some discussion on the elaboration/optimisation of fragments.
The pages are
Fragment-Based Screening
Building a Fragment Collection
Available Fragment Collections
Profiles of Fragment Collections
Fragment-Based Screening Published Hits
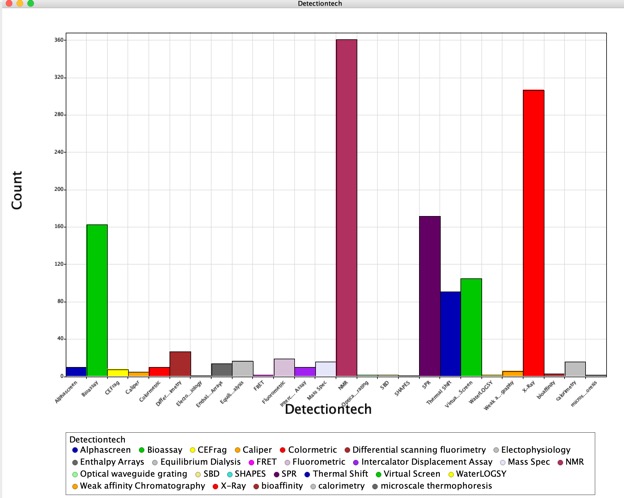
The published fragments contains details of fragments that have been reported as hits in the literature, this database now has over 1500 entries culled from over 310 publications directed at nearly 220 different molecular targets using 26 different detection technologies.
It could be argued that published fragment hits perhaps gives us an insight into the best fragments to include in library design.
BioBlocks
I've added a new entry on to the available fragments page, BioBlocks is a newcomer to the field of fragment collections. Whilst many collections are culled from available compound collections using calculated property filters (eg Rule of 3), BioBlocks have designed novel fragments and as such there is negligible overlap with other collections. One concern with bespoke fragments is that it is often a challenge to find related analogues for followup. The BioBlocks Comprehensive Fragment Library (CFL) is a subset generated from a >1 million member synthesizable virtual library, so follow up compounds can be generated using proven in house chemistry.
Practical Fragments Poll
The latest fragment-finding methods poll has been published on Practical Fragments.
The results underline the increase in the use of fragment based screening across the industry with 85% of the respondents now reporting that they actively use fragment screening. The technologies used to detect binding have also diversified with X-ray, NMR and SPR dominating. This mirrors my findings from published fragment hits. The choice of detection technology may be due to the additional structural information that X-Ray and NMR can offer.
I was delighted to see this comment,
For the first time we asked about use of literature to identify fragments, and nearly a third of respondents said they incorporate previously published fragments into their work. As the amount of publicly available information continues to increase it will be interesting to see whether this number grows.
I'll be updating the published fragment hits at the end of the year.
Website Update
I've spent some time over the last couple of weeks updating and adding new content to the Drug Discovery Resources section of the website.
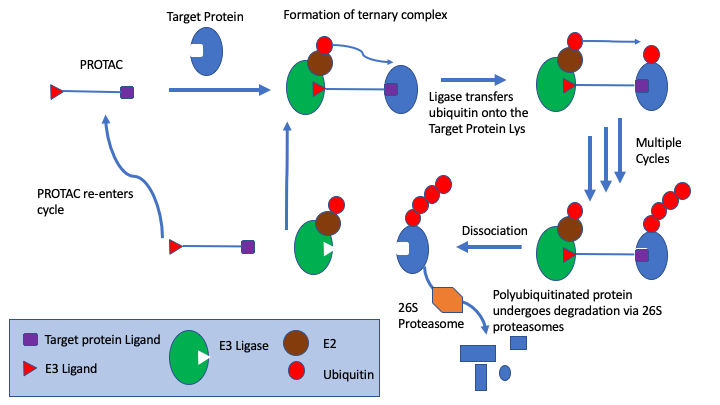
In particular, s drug targets become more challenging medicinal chemists are looking at alternatives to small molecule competitive inhibitors, the section on covalent inhibitors have been expanded and a new page on PROTACs has been added. PROTACs are bifunctional molecules that bind to the target protein and an E3 ligase, the simultaneous PROTAC binding of two proteins brings the target protein in close enough proximity for polyubiquitination by the E2 enzyme associated to the E3 ligase, which flags the target protein for degradation through the proteasome. This offers a powerful alternative to competitive inhibition.
The Probes & Drugs portal has been added to the chemical probes page, this is a public resource joining together focused libraries of bioactive compounds (probes, drugs, specific inhibitor sets etc.) with commercially available screening libraries.
The page describing commercial fragment screening libraries has been updated to include a couple of new additions and flagging some that seem to be unavailable, if I've missed any feel free to let me know.
The section on hERG has been updated with links to new references and details of hERGcentral.
hERGCentral: A Large Database to Store, Retrieve, and Analyze Compound-Human Ether-à-go-go Related Gene Channel Interactions to Facilitate Cardiotoxicity Assessment in Drug Development. The hERGCentral database hergcentral.org is based on experimental data obtained from a primary screen by electrophysiology against more than 300,000 structurally diverse compounds screened at 1 and 10uM.
Unfortunately the database appears to be no longer available. Whilst the supplementary information for the original publication does not contain the structures of the tested compounds it does reference the PubChem substance ID. I used these identifiers to download the structures of the >300,000 records and combined them with the experimental data provided in the Excel tables in the supplementary information. The complete dataset can be downloaded from the hERG page.
Small molecules can potentially bind to a variety of bimolecular targets and whilst counter-screening against a wide variety of targets is feasible it can be rather expensive and probably only realistic for when a compound has been identified as of particular interest. For this reason there is considerable interest in building computational models to predict potential interactions the page on predicting bioactivities has been expanded.
The section on bioisosteres also have a few new examples.
Drug Discovery Resources Updated
I've spent a little time updating the Drug Discovery Resources Section of the website. In particular:
- CYP interactions now includes details of published crystal structures and more information on known inhibitors
- I've added page on CYP1A2 from ChEMBL data and updated the CYP2D6 and CYP3A4 pages
- Updated the page on Aldehyde Oxidase
- Added new examples on the bioisosteres page
- Updated the Published Fragments section, including adding the overlay of all examples of Kinase fragment hits from the PDB.
- Added new examples to the Chemical Probes page
- Included more examples of halogen bonding to the Molecular Interactions page
Fragment Based Screening
I've made a few updates to the fragment-based screening approach to drug discovery pages.
Added a little on strategies for following the initial leads and also updated the details on drugs in the clinic discovered using fragment-based approaches.
Fragment Screening - XChem at Diamond
Fragment-based screening has become increasingly popular over the last 10 years and has proven to be a viable alternative to high-throughput screening. The appeal has been driven by several features
- “Fragment Space” is smaller than “Chemical Space” and can be more effectively probed with a relatively small library
- A million compounds cover only a small fraction of the suggested 1060 Chemical Space, whilst 2000 compounds can probe much of the 106 Fragment Space
- Protein requirements should be smaller
- Binding Efficiency for small molecules is likely to be higher
- Hit rates for Fragment-based screening appear to be higher, typically 3-10%.
Whilst there are a number of biophysical methods used for Fragment-based screening structural information is often a critical step in moving the programme forward. X-ray crystallography is a very powerful technology for use in converting a "hit" into a lead for drug discovery. However, the experimental overheads have historically been too high for it to be widely used for primary screening. The X-Chem project at Diamond aims to make the technology more widely accessible.
At Diamond beamline I04-1, the full X-ray screening experiment has now been implemented as a highly streamlined process, allowing up to 1000 compounds to be screened individually in less than a week (including 36 hours' unattended beamtime). The process covers soaking, harvesting, automatic data collection, and data analysis; fragment libraries are available, though users can bring their own.
An overview of the process is available here in practice, users must generate the crystals in their home lab, and are required to come and perform soaking and harvesting themselves. Users do not need to be present for the X-ray data collection when data is collected automatically. Data analysis builds on the existing automatic data processing, and they have developed tools to streamline density interpretation and refinement (PanDDA and XChemExplorer). Use of these tools at Diamond is optional but highly recommended.
Whilst users are free to bring their own libraries a number of libraries are also available these include the Maybridge 1000, Edelris fragments, and Diamond-SGC Poised Library (DSPL), a fragment library designed to allow rapid, cheap follow-up synthesis to provide quick SAR data. The calculated physicochemical properties of the DSPL fragment collection are shown below.
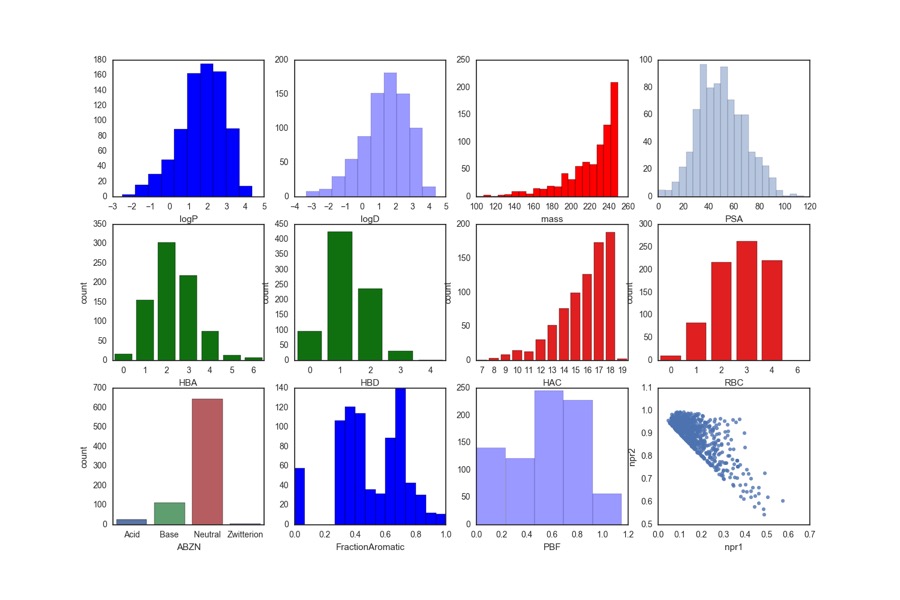
For more details on the design of the library O. B. Cox, K. Krojer, P. Collins, O. Monteiro, R. Talon, A. Bradley, O. Fedorov, J. Amin, B. D. Marsden, J. Spencer, F. von Delft, P. E. Brennan, A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem. Sci.,2016. 7: p. 2322-2330 DOI.
There is more information in this podcast https://www.ndm.ox.ac.uk/frank-von-delft-x-rays-for-drug-discovery
Fragments 2017
Fragment-based screening is now a well established methodology for the identification of leads for drug discovery and the aim of the 6th RSC-BMCS Fragment-based Drug Discovery meeting will be to continue the focus on case studies in Fragment-based Drug Discovery that have delivered compounds to late stage medicinal chemistry, preclinical or clinical programmes.
6th RSC-BMCS Fragment-based Drug Discovery meeting
Sunday to Tuesday, 5th to 7th March 2017
at Parkhotel Schönbrunn, Vienna, Austria
Full details and registration are online.
Two conferences that might be of interest
| Event | FBLD 2016 (Fragment-based Lead Discovery Conference 2016) |
|---|---|
| Place | Cambridge, Massachusetts, USA |
| Dates | 9th-12th October 2016 |
| Website | http://www.ysbl.york.ac.uk/fbld/2016/ |
| Poster abstracts | Closing date is 30th September |
| Event | Fragments 2017 - 6th RSC-BMCS Fragment-based Drug Discovery meeting |
| Dates | Sunday to Tuesday, 5th to 7th March 2017 |
| Place | Parkhotel Schönbrunn, Vienna, Austria |
| Websites | http://www.maggichurchouseevents.,co.uk/bmcs |
| also | http://www.rsc.org/events/detail/23352/fragments-2017-7th-rsc-bmcs-fragment-based-drug-discovery-meeting |
Fragment based screening
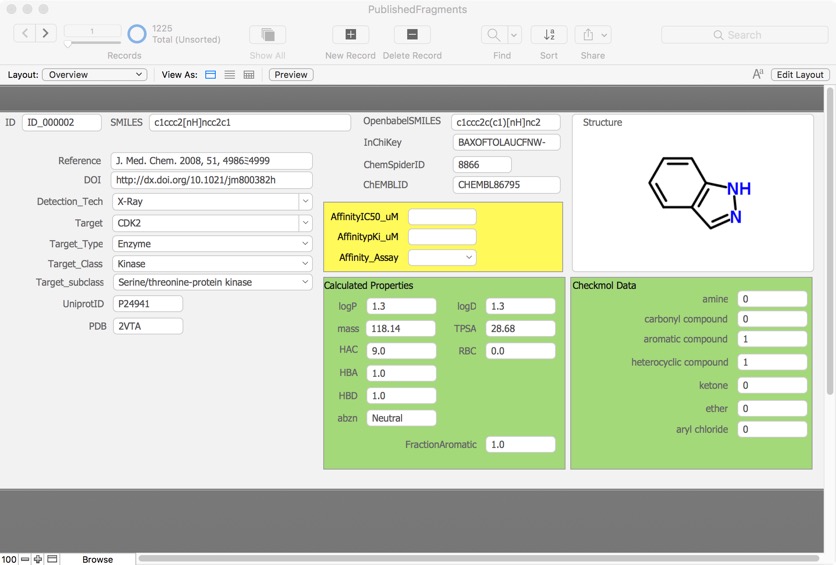
I’ve just updated some of the fragment based screening pages, in particular I’ve updated the section on Published fragment Hits. The database now contains 1216 entries culled from over 240 publications directed at nearly 174 different molecular targets using 26 different detection technologies.
I also noticed that a fragment library I was helped design is now commercially available, The Selcia Fragment Library was designed to have broad applicability and chemical tractability. It is also one of the few libraries where solubility has been confirmed experimentally. The profile of the library is included in the fragment library profiles.
Fragment Screening at Diamond
I'm in the process of updating the fragment-based screening section of the Drug Discovery Resources and I came across this news article from the Diamond Light Source is the UK’s synchrotron.
At Diamond beamline I04-1, the full X-ray screening experiment has now been implemented as a highly streamlined process, allowing up to 500 crystals to be soaked and harvested in a day, and collected in 24 hours beamtime. The process covers soaking, harvesting, automatic data collection, and data analysis; and fragment libraries are available, or users can bring their own.
This is available to both academic and commercial users but the application process is different.
If you are interested there is a very useful checklist that should simplify the process.
The facility is based at beamline I04-1 and nearby Lab 36, where the soaking and harvesting is performed.
In practice, the experiment will span a few days and even multiple visits to establish crystals' suitability. Users must generate the crystals in their home lab, and are required to come and perform soaking and harvesting themselves: multi-day Lab Visits will be scheduled separate from normal beamline time. In contrast, users do not need to be present for the X-ray data collection, although they are asked to help monitor (remotely) the automated collection when it occurs. A local contact will be assigned, same as for beamtime.
In practice, the first steps to unsure reproducibility are iterative and require a few dozen crystals, and in difficult cases even several Lab Visits; but associated diffraction testing will be fitted in during the Lab Visit where possible. The final "Full run" soaking and harvesting will be scheduled once the soaking protocol is confirmed (in favourable cases during the same Lab Visit).
Data analysis builds on the existing automatic data processing, and we are developing tools to streamline density interpretation and refinement, analysis and presentation of hit results, and depositing hit structures. Use of these tools is optional, but feedback valued: they will be deployed on Diamond's compute environment as they become available.
The Diamond fragment collection is actively evolving from the original Maybridge fragment collection, on the one hand to eliminate compounds that are poorly soluble, unstable or systematically kill crystals. On the other hand it is being expanded with synthesis-ready compounds.
Caveat emptor
A paper entitled Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold appeared in J Med Chem recently DOI, in which they describe the promiscuous nature of 2-aminothiazoles in screens.
Exemplified by 4-phenylthiazol-2-amine being identified as a hit in 14/14 screens against a diverse range of protein targets, suggesting that this scaffold is a poor starting point for fragment-based drug discovery
I thought I'd check how often this substructure appears in the published fragments database, indeed currently 43 of the 903 published fragments contain this substructure. Further investigation identifies a total of 63 amino-substituted 5-membered heterocycles, and there are 167 fragments in which there is an amino group on an aromatic ring (mainly heterocycles).
It should also be noted however that there are 64 structures in the DrugBank database that also contain a 2-aminothiazole, so whilst promiscuous they can be developed into drugs.
So whether they are privileged structures or troublesome promiscuous hits is probably in the eye of the beholder, caveat emptor.
Fragment sized drugs
As someone who regularly reads Derek Lowe’s “In the Pipeline” blog I was taken with the post on The Smallest Drugs in which he highlighted the structures using the arbitrary cutoffs
the molecular weight cutoff was set, arbitrarily, at aspirin's 180. I excluded the inhaled anaesthetics, only allowing things that are oils or solids in their form of use. As a small-molecule organic chemist, I only allowed organic compounds - lithium and so on are for another category.
An interesting selection but I thought it might be interesting to profile the calculated properties, I used the DrugBank Database too ensure I got a more comprehensive dataset and then calculated properties as I have done for the Fragment collections. The results are shown below. Probably the most notable feature is the number that contain ionisable groups, over 60% of the molecules would be predicted to be ionised at physiological pH (note however it does include a couple of natural amino acids). Around 50% contain an aromatic ring (of which 2/3 are heterocycles). There are a couple of structures with more 3D shape (Memantine) but in general they would be classified as disc or rod-like. In general the results don’t look too dissimilar to the Published Fragment Hits.
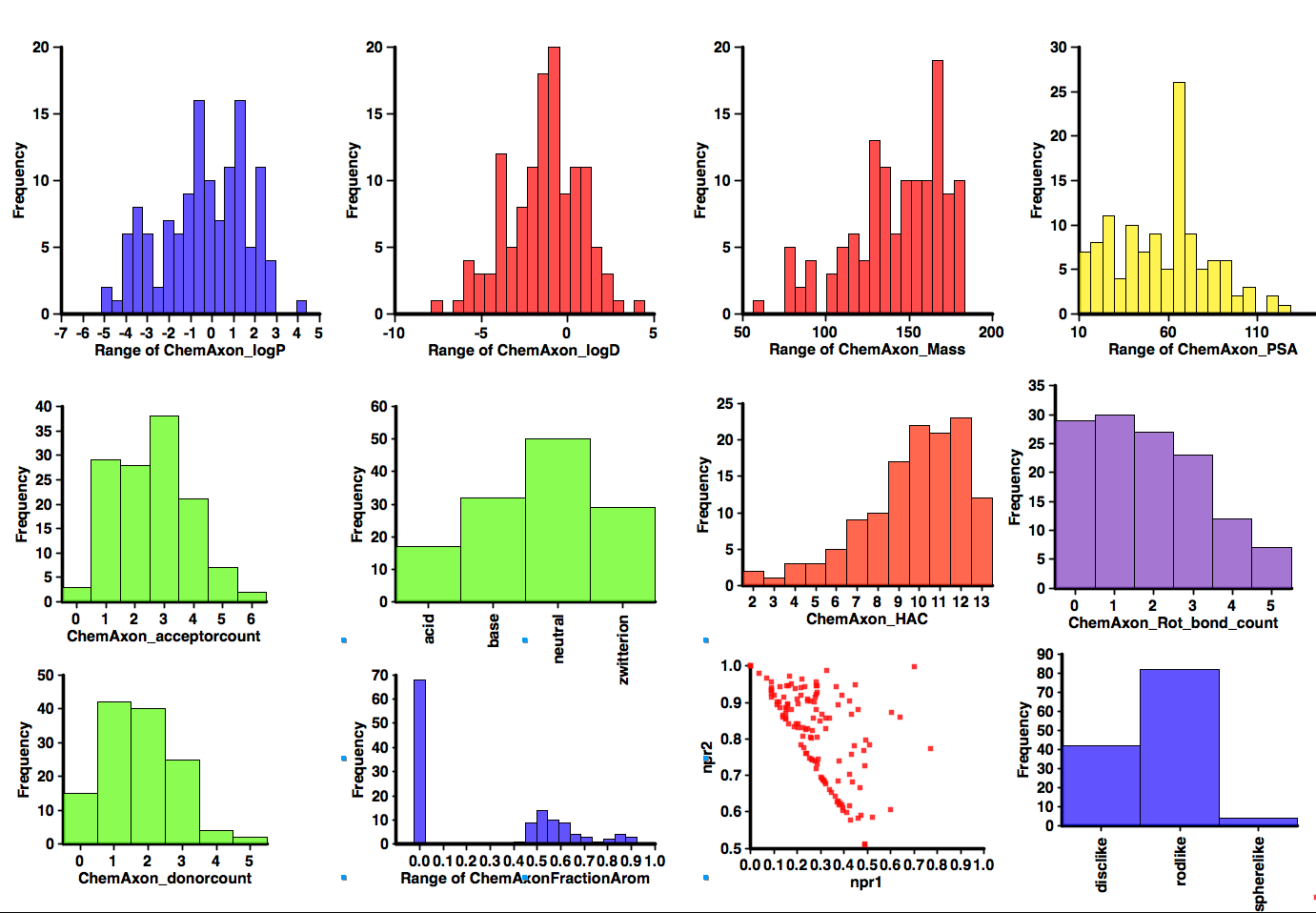
Published Fragments
I’ve updated the page on published fragments, the dataset now includes over 800 published fragments hits abstracted from over 200 publications directed at nearly 130 different molecular targets using 22 different detection technologies and might be expected to give some insight into the type of compounds that appear as hits. With the caveat that the dataset only includes information that has been published.
Drug Discovery Resources Updates
I’ve made a couple of updates to the Drug Discovery Resources pages. In particular I’ve updated the Published fragments Hits to include more examples, details of “promiscuous” compounds and summary of detection technologies and the targets explored. I’ve also updated the Aspartic Protease inhibitors page.
As ever comments and/or suggestions very welcome.
New fragment libraries
It is interesting to see how commercial fragment libraries are starting to evolve, from simple molecular weight cuts of available chemicals to more careful selection based on physicochemical properties. We now see several interesting design strategies being adopted.
Those based on a screening technology such as the LifeChemicals Fluorine-based library to support 19F NMR-based fragment screening, and the Maybridge Bromo-Fragment Collection a collection of over 1500 bromine containing Maybridge fragments constructed as an aid to X-ray based fragment screening.
Other libraries are designed for specific targets
OTAVA offers you new Chelator Fragment Library that comprises 575 compounds in total, Chelators demonstrate binding affinities suitable for FBLD screening and provide a diverse range of molecular platforms from which to develop lead compounds. Also, the propensity for chelators to bind metal ions allows for better prediction of their probable binding position within a protein active site in the absence of experimental structural data of the complex.
Many attractive drug targets contain a free sulfhydryl group in the active site that confounds functional HTS assays due to its facile, non-specific oxidation leading to target inhibition. AnCore have developed a Targeted Covalent Inhibitor fragment library (TCI-Frag™) containing 100+ Rule-of-3 compliant fragments are conjugated with mildly reactive functionalities. The BIONET CNS Fragment Library is a focused library containing 700 Fragments selected for their suitability for Fragment Based Lead Discovery in the areas of CNS drug discovery and Universal target classes.
I’ve updated the Fragment Collections page
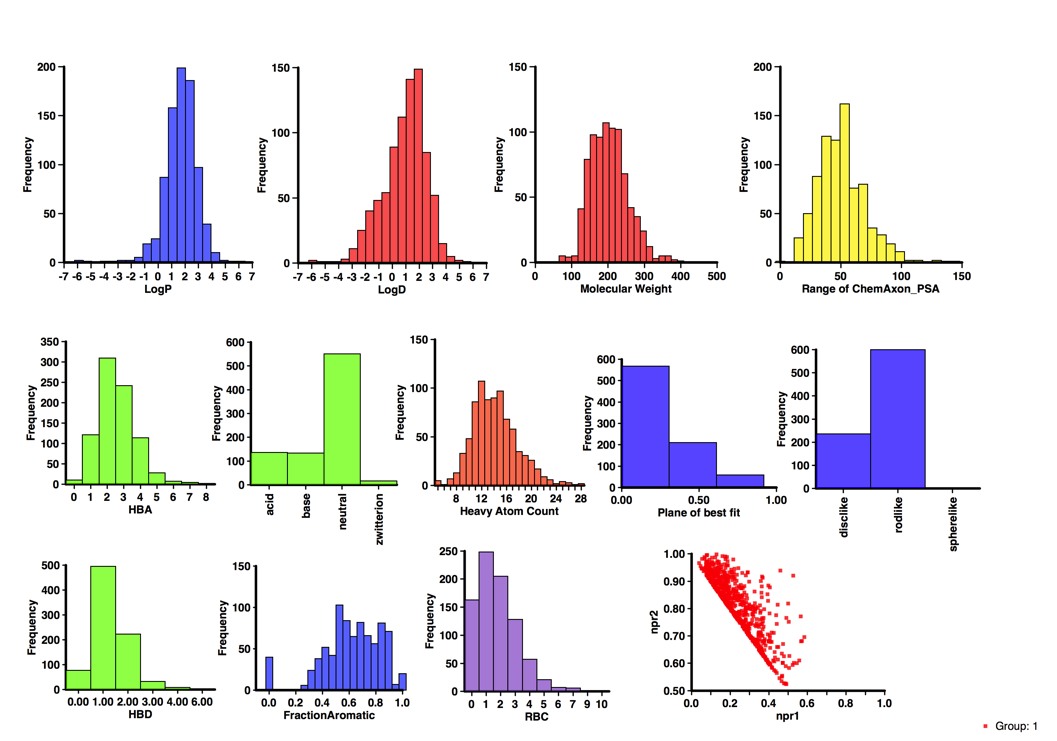
Published Fragment Hits
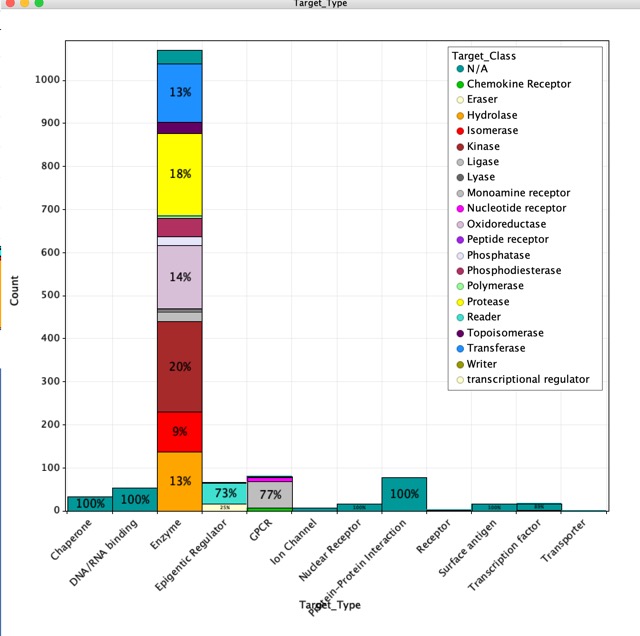
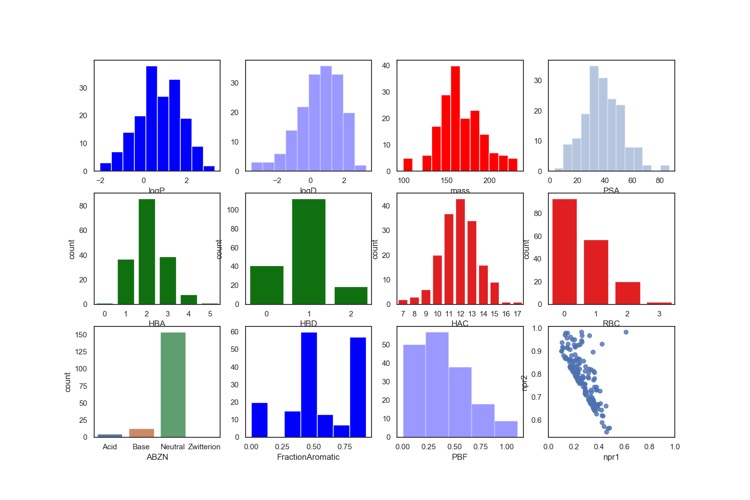
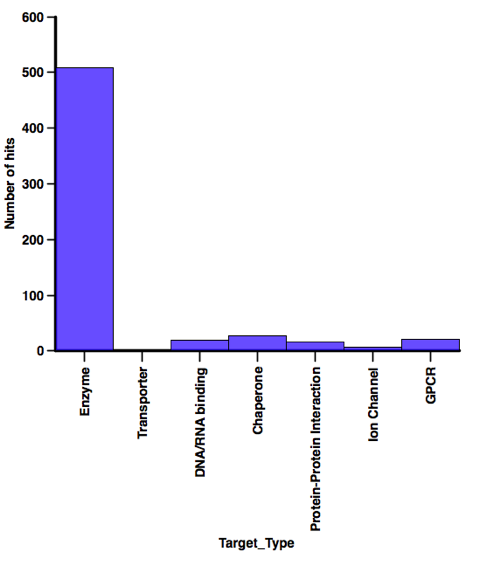
I’ve continued to collect details of fragment based screening hits that have been reported in the literature. There are now over 600 hits reported for 113 different targets culled from over 160 publications. I’ll update the calculated properties for those compounds in due course. I was interested in seeing if the physicochemical profiles are different depending on the type of target, however as the plot below shows, the majority of those hits have been identified against enzyme targets so I think I’ll need more data before any meaningful conclusions can be made.
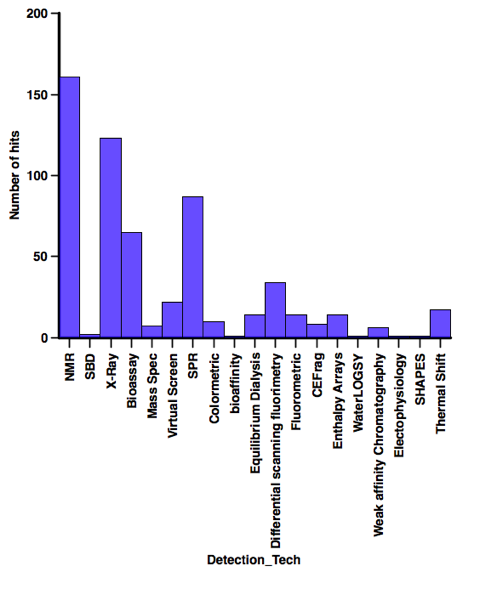
In contrast when looking at the screening technology used a variety of technologies have afforded a substantial number of hits, when I’ve abstracted the latest batch of papers I’ll have a look at the profiles of the compounds identified using each technology.
Finding the data is getting more of a challenge, it seems as fragment screening becomes more mainstream it is often not mentioned in the title or abstract. So if you have recently published a relevant paper if you could send me the reference or even a pdf I’d be very grateful.
Shape distribution for fragment collections
I’ve now updated the physicochemical property profiles of all the fragment collections I have access to, including the categorisation into rod-, disc- or sphere-like shapes I described last week.
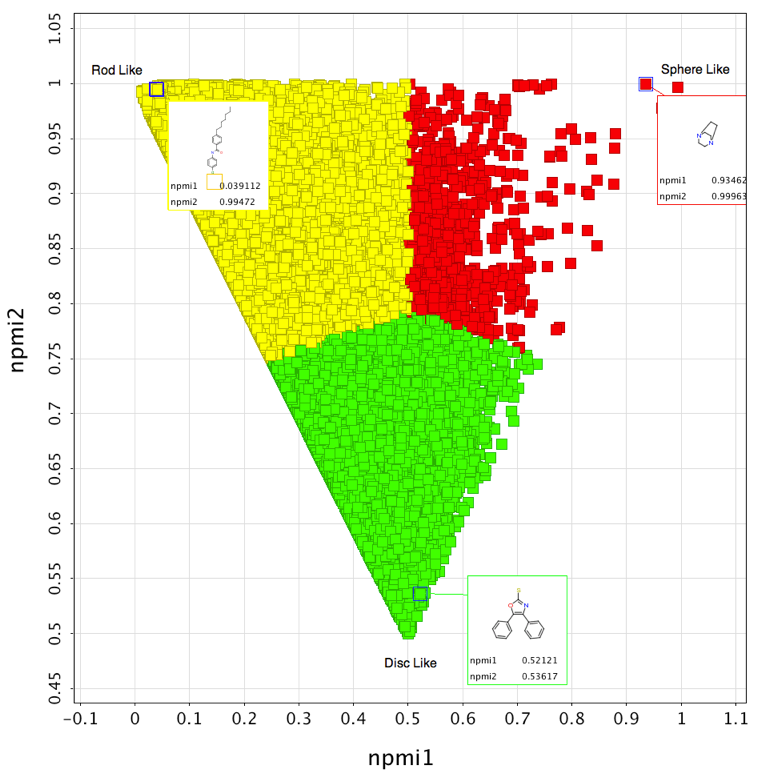
I thought it might be interesting to generate a plot of all 170,000 fragments to look at the distribution. I actually viewed the results in Vortex as shown below. This tool makes it easy to colour by “shape” and also allows me to highlight a few structures that appear at the extremism of the plot.
3D Shape distributions of Compound Collections
I recently updated the fragment collections page this included updating the physicochemical property profiles adding npmi (Normalized ratio of principle moments of inertia) as described by Sauer WH, Schwarz MK (2003) Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J Chem Inf Comput Sci 43:987–10030. DOI As the image below shows this gives a view of the shape of the molecules as to whether they are rod, disk or sphere like.
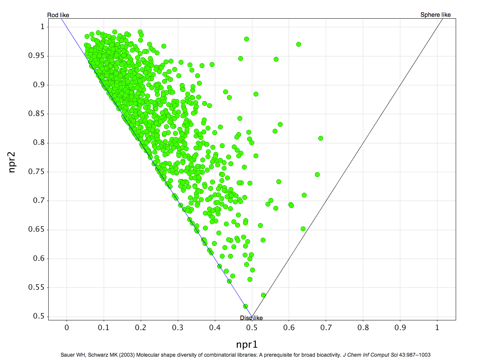
Whilst this works very well for individual compounds or small libraries the plot becomes a blur of overlapping points for larger collections and it is not really possible to compare collections. Whilst it may be possible to generate a single number as the “average” of each collection I’m not sure how useful it would be. So with help from Matt I decided to divide the plot into three sections as shown below.
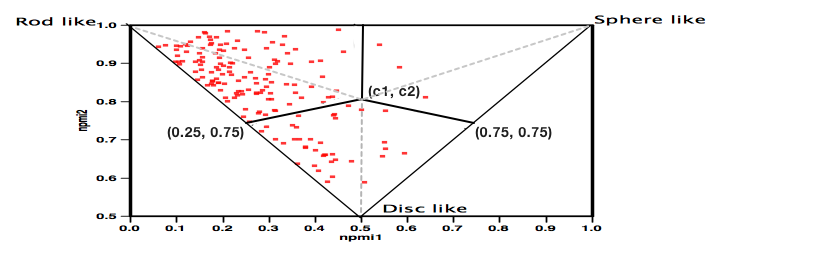
The centre point (c1, c2) was calculated using ( 0.5, (2sqrt(0.5) + 0.5)/(2sqrt(0.5) + 1) ) which is about (0.5, 0.793).
Each of the points in the plot was then assigned to a category using:
If a point is below both lines then: (0.5 - 0.25) * (npmi2 - 0.75) - (0.793 - 0.75) * (npmi1 - 0.25) < 0 and (0.5 - 0.75) * (npmi2 - 0.75) - (0.793 - 0.75) * (npmi1 - 0.75) > 0 then it is disc-like.
If not, then it is rod-like if npmi1 < 0.5 and sphere-like if npmi1 > 0.5.
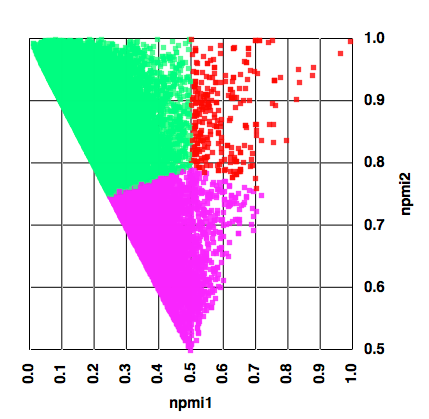
The result for a 35,000 compound collection are shown below, with the points colour-coded by the assigned category.
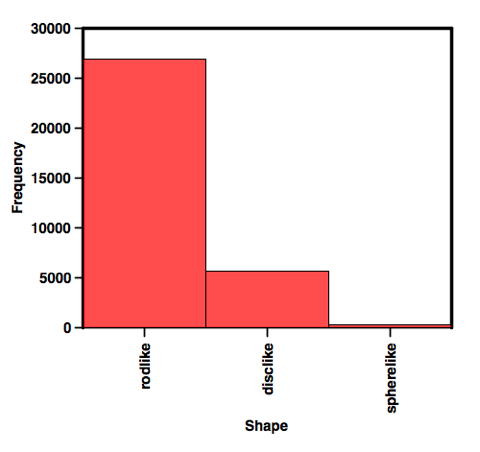
We can then create a categorical plot as shown below.
I plan to update all the physicochemical profiles of all the fragment collections next week.
3D Fragment Consortium
I just added the dataset from the 3D Fragment Library Consortium.
The 3D Fragment Consortium brings together UK-based not-for-profit drug discovery institutes and academic groups, working in partnership to build a collection of chemically diverse molecules with a particular focus on fragments that incorporate 3D structure. The consortium is looking to collaborate with other research groups to expand the collection and make it available for screening against new biological targets to help kick-start hit discovery programmes and provide a foundation for a vibrant pre-competitive drug discovery network across the UK. The 3D Fragment Consortium has identified a foundation library of 170 fragments to commence their screening activities.
I’ve added this fragment library profile to the 22 other collections previously calculated:
Comparison of all collections, and calculated physiochemical profiles.
It is obviously early days yet but it will be interesting to see how this develops.
How are fragments optimised?
A recent paper J. Med. Chem. 2013, 56, 2478−2486 DOI looked at the different ways that the initial fragments were subsequently optimised, looking at a variety of physicochemical properties and ligand efficiency indices.
I’ve updated the known fragments page to include some of their conclusions.
Known Fragment Hits
I’ve just updated the section describing fragment hits reported in the literature.The dataset now has >500 entries culled from 150 publications directed at nearly 100 different molecular targets using 18 different detection technologies and might be expected to give some insight into the type of compounds that appear as hits.
I’ve calculated a number of physicochemical descriptors and identified fragments that have appeared as hits against multiple targets.
Fragment Pages updated
I’ve just updated the section on fragment libraries, I’ve added a couple of new vendors and updated the existing vendors, there are now over 160,000 fragments available from commercial suppliers. I’ve recalculated the identity and similarity matrix. I’ve also updated the physicochemical property profiles and added npmi (Normalized ratio of principle moments of inertia) as described by Sauer WH, Schwarz MK (2003) Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J Chem Inf Comput Sci 43:987–10030. DOI As the image below shows this gives a view of the shape of the molecules as to whether they are rod, disk or sphere like, it is included with all the other calculated properties.
I notice that a couple of the vendor with very large fragment collections now sell relatively small subsets, underlining the fact that a library of 2000 fragments is usually sufficient as a screening set. Access to the larger fragment space is only really needed when you come to explore the hits.
Learning from our mistakes: The ‘unknown knowns’ in fragment screening
Whilst fragment-based screening has been around for a while there are still groups that are new to the area. This invaluable paper provides an insight into the pit-falls that await the unwary scientist. Absolutely essential reading
In the past 15 years, fragment-based lead discovery (FBLD) has been adopted widely throughout academia and industry. The approach entails discovering very small molecular fragments and growing, merging, or linking them to produce drug leads. Because the affinities of the initial fragments are often low, detection methods are pushed to their limits, leading to a variety of artifacts, false positives, and false negatives that too often go unrecognized. This Digest discusses some of these problems and offers suggestions to avoid them. Although the primary focus is on FBLD, many of the lessons also apply to more established approaches such as high-throughput screening.
Learning from our mistakes: The ‘unknown knowns’ in fragment screening DOI
Published Fragment hits
Whilst there are a variety of techniques to measure the properties or diversity of fragment libraries it is interesting to look at the profiles of compounds that actually appear as hits in fragment-based screening campaigns. I’ve been compiling a database of compounds that have been reported as hits in the literature, this database now has >500 entries culled from 150 publications directed at nearly 100 different molecular targets using 18 different detection technologies and might be expected to give some insight into the type of compounds that appear as hits.
Suggested Books
I’ve just updated the list of suggested books.
Included books on bioisosteres and fragment-based screening.
Fragment Collections
I’ve just updated the page containing the profiles of commercial fragment collections.
Fragment library design: the evolution of fragment-based lead discovery
Latest Publication
Fragment library design: the evolution of fragment-based lead discovery
By Dr E. Zartler, Dr C. Swain & S. Pearce
Drug Discovery World Winter 2012/13
With the growing need to streamline the drug discovery process, screening against fragment libraries rather than drug-like molecules has become increasingly adopted as an integral part of many drug discovery programmes. However, success depends on the quality of the fragment library, and many factors dictate quality.
Fragments in the Clinic Updated
Practical Fragments has updated the its list of fragment-derived compounds in the clinic. There is now one approved, the B-Raf(V600E) inhibitor Vemurafenib, eleven apparently in Phase 2 or 3 and fourteen reportedly in Phase 1.
Drug Discovery Resources Update
I’ve updated the Drug Discovery Resources Pages over the Christmas Break. In particular I’ve updated the Fragment Based Screening section and added a page on building a fragment collection. I’ve also updated the section on CYP interactions, expanding the Induction section.
Updated Frament Screening Pages
I’ve updated the fragment-based screening pages, added new screening technologies, added a couple of new vendors and updated the fragment collection profiles based on the current fragment collections that vendors have provided.
Fragment methods in drug discovery: the potential grows
An interesting set of articles focussed on Fragment methods published in Drug Discovery Today.
Fragment methods have now become well established within the repertoire of drug discovery technologies used within the pharma and biotech industries. Success has been repeatedly demonstrated in the application of fragment methods as the basis for the discovery of drug candidates with attractive physicochemical properties for soluble protein targets….In this issue of Drug Discovery Today, Editor’s Choice highlights four recent papers that consider how to make the best use of fragments both in terms of their optimisation and their application more broadly in drug discovery. The papers featured are all available as free downloads, so please have a look at them; I’m sure that you will find them interesting and thought provoking.
Fragment Screening Publication
http://dx.doi.org/10.1177/1087057112445785
Fragment Profiles Updated
Fragment Screening Pages Updated
I’ve just updated the pages devoted to fragment-based screening
Fragment Collections
Fragment Collection Profiles
Fragment-Based Screening
Known Fragment Hits
Many thanks to all who provided details of their fragment collections.
Updated Fragment Collections
I’ve just updated two pages.
Fragment Collections on which I’ve added a couple of additional suppliers, and Fragment collection profiles on which I’ve added the profile of the TimTec collection and updated the collections from the existing vendors. It seems that as the collections evolve more of the collections are starting to look like they were designed rather than simply applying cutoffs (MWt etc) to an existing sample collection. We are starting to see less aromatic compounds and more consideration to the 3D structure.
Updated Fragment Collection Profiles
Several companies have kindly sent me updated copies of the structure files for their fragment collections.
I’ve updated the analysis of the collections and added a couple more properties, by calculating the most acid/basic pKa I’ve catagorised the fragments into acid/base/neutral/zwitterion. I’ve also calculated the fraction of aromatic atoms (number of aromatic/number heavy atoms), one comment I’ve heard about some collections is that they contain too many aromatic compounds and have issues with solubility.
Full details are on the Fragment Collection Profiles page.
Fragment Screening
I’ve just updated the page on fragment collection profiles
Full details are on the Fragment Collection Profiles page.
Fragment Screening
Just completed a project to help design a fragment library for Selcia to be used with their proprietary fragment screening technology CEFrag.
A poster will be presented at the FBLD2010 meeting in Philadelphia in October
Updates
I’ve added a sections on fragment-based screening, solubility and updated the section on metabolism.