
Antiviral Competition opens Jan 13th
As part of its open science mission, the ASAP Discovery Consortium is conducting a computational methods competition encompassing several modalities critical to small molecule drug discovery. This competition will be run in collaboration with OpenADMET, which is a new ARPA-H funded project under the Open Molecular Software Foundation (OMSF).
This competition will be composed of three sub-challenges:
Ligand Poses: ASAP has produced a large volume of X-ray crystallography data over its years of operation. Along this trajectory, SARS-CoV-2 Mpro was structurally enabled much earlier than MERS-CoV. This sub-challenge will recreate that situation. Given a training set of SARS-CoV-2 Mpro X-ray structures, participants will be asked to predict poses of a test set of compounds for MERS-CoV Mpro. The crystallography experiments for this sub-challenge were performed by the University of Oxford and Diamond Light Source. See here for the crystallography conditions.
Potency: Given a training set of dose-response fluorescence potency data for both targets (SARS and MERS Mpro), participants will be challenged to predict potencies for a blind set of compounds for both targets. The assays for this sub-challenge were performed by the Weizmann Institute of Science. See here for the experimental conditions.
ADMET: This sub-challenge will consist of multiple ADMET endpoints. Participants will receive training data for all endpoints and will be asked to predict the same endpoints for a blind set of compounds. The assays for this sub-challenge were performed by Bienta.
Full details and preliminary data are available online. https://polarishub.io/blog/antiviral-competition.
CCDC: Curated Data Set of Protein Structures
Fantastic news from the Cambridge Crystallographic Data Centre (CCDC), a curated data set of protein structures from the Protein Data Bank (PDB) with predicted hydrogen positions is now available for download. The dataset is taken from the Protein Data Bank (PDB) and has the positions of hydrogens accurately computed, this provides a comprehensive snapshot of protein cavities in the PDB, identifying potential binding sites for small molecules with accurately predicted hydrogen positions for all components.
The news article is here https://www.ccdc.cam.ac.uk/discover/blog/accelerating-drug-discovery-with-the-ccdc-aws-and-intel/.
This large subset of the protein data bank which has be processed using the CCDC's protonation workflow so that reasonable proton positions have been modelled can be downloaded here.
https://www.ccdc.cam.ac.uk/support-and-resources/downloads/.
Drug design data sets for testing computational tools
One of the challenges when building novel tools to aid drug discovery is identifying high quality datasets that can be used to test new tools. This is why the D3R datasets are so valuable https://drugdesigndata.org.
These datasets are available from BindingDB and include a variety of important protein targets.
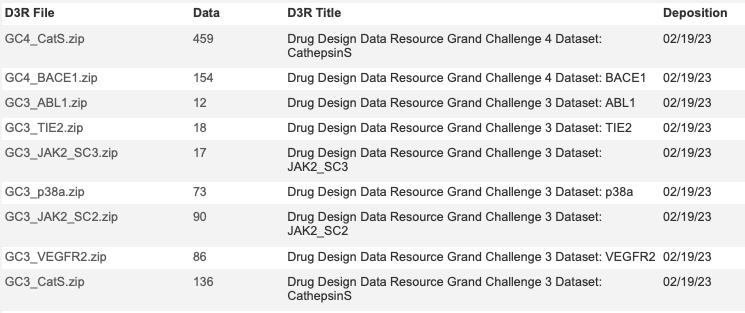
The targets include CathepsinS, BACE1, ABL1) and JAK2.
Major user experience update in AlphaFold Database
Just saw this.
The AlphaFold Protein Structure Database, a result of a collaborative effort between Google DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI), has released an exciting update to its web pages, providing users with an enhanced experience. This update marks a significant step in facilitating the use of AlphaFold structure data.
One of the most interesting updates are the improvements to the 3D viewer Mol*.
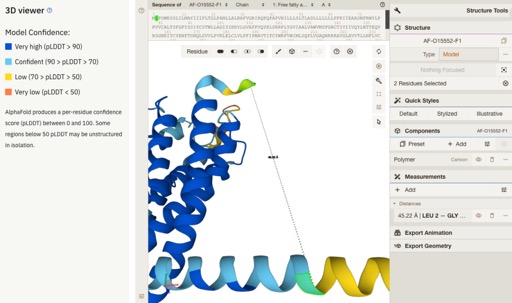
Full details are here https://www.ebi.ac.uk/about/news/updates-from-data-resources/alphafold-database-ux-update/.
Predicting sites of metabolism
I updated the page on predicting metabolism
https://www.cambridgemedchemconsulting.com/resources/ADME/predicting_metabolism.html.
3rd In Silico Toxicology Conference
The 3rd In Silico Toxicology Conference, supported by the British Toxicology Society (BTS), the Royal Society of Chemistry (RSC) CICAG Group, Lhasa Ltd., and the Cambridge Alliance on Medicines Safety (CAMS) will take place online on 29 September 2022; attendance is free and open to everyone interested.
Topics will include In Silico Toxicology Consortia, Cell Painting, Gene Expression Data, Biomarkers, Interpreting Neural Networks, Drug-Induced Liver Injury/DILI, Skin Sensitization, Animal Histopathology Data, Species Concordance, In Vivo Pharmacokinetics (PK), Molecular Initiating Events (MIEs), Chemicals, Pharma, Food, Read-Across, ... and beyond (see website for the full programme and registration).
AI4Proteins videos now online
On June 16/17 2021 RSC CICAG and AI3D held a joint meeting on Protein Structure Prediction. The full lineup of speakers, titles and abstracts can be found here.
Session 1: Session Chair: Professor Jeremy Frey (University of Southampton)
An AI solution to the protein folding problem: what is it, how did it happen, and some implications Professor John Moult (University of Maryland)
Session 2: Session Chair: Dr Melanie Vollmar (Diamond)
So you predicted a protein structure – What now? Dr Thomas Steinbrecher (Schrödinger)
Deep Learning enhanced prediction of protein structure and dynamics Dr Martina Audagnotto (AstraZeneca)
Fireflies-Lévy Flights algorithm for peptides conformational optimization Dr Zied Hosni (University of Sheffield)
Session 3: Session Chair: Dr Chris Swain (Cambridge MedChem Consulting)
How good are protein structure prediction methods at predicting folding pathways? Mr Carlos Outeiral Rubiera (University of Oxford)
Protein-Ligand Structure Prediction for GPCR Drug Design Dr Chris De Graaf (Sosei Heptares)
Session 4: Session Chair: Dr Márton Vass
Using icospherical input data in machine learning on the protein-binding problem Dr Ella Gale (University of Bristol)
Biological sequence design with machine learning Professor Debora Marks (Harvard University)
Session 5: Session Chair: Dr Simone Fulle (Novo Nordisk)
Lessons learned from generative models of biological sequences Professor Aleksej Zelezniak (Chalmers University of Technology)
DeepDock: a deep learning approach to predict ligand binding conformations Dr Oscar Méndez-Lucio (Janssen Pharmaceuticals)
Finding new in silico-based therapeutic strategies for IAHSP Dr Matteo Rossi Sebastiano (University of Turin)
Session 6: Session Chair: Professor Jonathan Goodman (University of Cambridge)
Designing molecular models by machine learning and experimental data Professor Cecilia Clementi (Freie Universität Berlin)
The “almost druggable” genome Professor Tudor Oprea (University of New Mexico)
Session 7: Session Chair: Dr Lucy Colwell (University of Cambridge)
General Effects of AI on Drug Discovery Dr Derek Lowe (Novartis)
Open Access Data: A Cornerstone for Artificial Intelligence Approaches to Protein Structure Prediction Professor Stephen Burley (RCSB PDB, Rutgers University, UCSD)
The videos of the presentations are now available on YouTube and you can access the playlist here https://www.youtube.com/playlist?list=PLBQwbn0mPhvWyTLnN6eFsbIwb5FByrs.
For those wanting a hype free insight into the impact AI might make on Drug Discovery then the presentation by Derek Lowe is well worth watching.
Computational Prediction of covalent Inhibitors
Covalent Inhibitors are an increasingly important class of therapeutic agents.
A computational pipeline has been described by the London lab to predict suggest covalent analogs of non-covalent ligands DOI.
Designing covalent inhibitors is increasingly important, although it remains challenging. Here, we present covalentizer, a computational pipeline for identifying irreversible inhibitors based on structures of targets with non-covalent binders. Through covalent docking of tailored focused libraries, we identify candidates that can bind covalently to a nearby cysteine while preserving the interactions of the original molecule. We found ∼11,000 cysteines proximal to a ligand across 8,386 complexes in the PDB. Of these, the protocol identified 1,553 structures with covalent predictions. In a prospective evaluation, five out of nine predicted covalent kinase inhibitors showed half-maximal inhibitory concentration (IC50) values between 155 nM and 4.5 μM. Application against an existing SARS-CoV Mpro reversible inhibitor led to an acrylamide inhibitor series with low micromolar IC50 values against SARS-CoV-2 Mpro. The docking was validated by 12 co-crystal structures. Together these examples hint at the vast number of covalent inhibitors accessible through our protocol.
RDKit was used for 2D molecular handling, conformation generation and RMSD calculation. RDKit: Open-source cheminformatics; version 2018.09.3; RDKit.org. Marvin was used in the process of preparing the molecules for docking, Marvin 17.21.0, ChemAxon (https://www.chemaxon.com). OpenBabel (http:// openbabel.org/wiki/Main_Page) was used to switch between molecular file formats. DOCKovalent (London et al., 2014) was used for virtual covalent docking. The Covalentizer code is available at https://github.com/LondonLab/Covalentizer.
The Polypharmacology Browser PPB2
Off-target activity is often ignored and might only be uncovered relatively late in the drug discovery program. Whilst broad spectrum screening is available it can be rather expensive. Predicting potential off-target activities is an attractive approach and this paper describes the development of a prediction tool using nearest neighbours combined with machine learning.
The Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning DOI
To build PPB2 we collected a bioactivity dataset of all compounds having at least IC50 < 10 uM on a single protein target in ChEMBL22 considering only high confidence data points as annotated in ChEMBL and only targets for which at least 10 compounds were documented
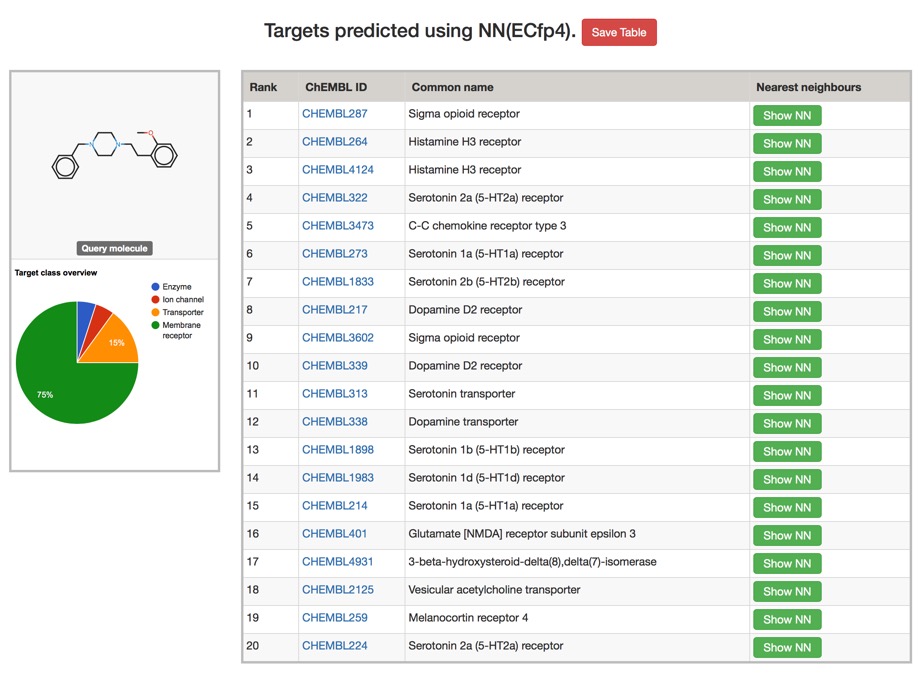
You can try it out here PPB2., depending on the model chosen the results are calculated in a couple of minutes, but don't post your proprietary molecules. Typical results are shown below, clicking on the green "Show NN" button shows the most similar structures.
Pitfalls to avoid when building a Computational Therapeutics Company
Everyday I seem to hear about another tech company moving into healthcare, whilst I'm certain that Artificial Intelligence and Machine Learning has the potential to make a significant impact this advice should be compulsory reading for all involved.
How many compounds do you select from virtual screening?
Whilst high-throughput screening (HTS) has been the starting point for many successful drug discovery programs the cost of screening, the accessibility of a large diverse sample collection, or throughput of the primary assay may preclude HTS as a starting point and identification of a smaller selection of compounds with a higher probability of being a hit may be desired. Directed or Virtual screening is a computational technique used in drug discovery research designed to identify potential hits for evaluation in primary assays. It involves the rapid in silico assessment of large libraries of chemical structures in order to identify those structures that most likely to be active against a drug target. The key question is then how many molecules do you select from your virtual screen?
Whilst virtual screening is certainly less expensive than high-throughput screening it is not free, even an in house academic cluster has an overhead (probably equating to > $10,000 per virtual screen). So with that investment how much would you invest in actual compounds?
Virtual Screening Pages Updated
I've updated the pages describing virtual screening, in particular the docking section.
Computational Tools Updated
I've updated the computational tools page in the Drug Discovery Resources.
Predicting bioactivities
Small molecules can potentially bind to a variety of bimolecular targets and whilst counter-screening against a wide variety of targets is feasible it can be rather expensive and probably only realistic for when a compound has been identified as of particular interest. For this reason there is considerable interest in building computational models to predict potential interactions. With the advent of large data sets of well annotated biological activity such as ChEMBL and BindingDB this has become possible.
These predictions may aid understanding of molecular mechanisms underlying the molecules bioactivity and predicting potential side effects or cross-reactivity.
A variety of options are summarised on this page.
Predicting Sites of Metabolism page updated
I've updated the Predicting sites of metabolism page.
Open Source Molecular Modeling
I’ve updated the Computational chemistry page to include a recent excellent publication, Open Source Molecular Modeling DOI a review that categorizes, enumerate, and describe available open source software packages for molecular modeling and computational chemistry.
There is also an online database https://opensourcemolecularmodeling.github.io that covers most aspects of computational drug discovery
Methods
Development Activity
Usage Activity
Cheminformatics
Toolkits
Standalone Programs
Graphical Development Environments
Visualization
2D Desktop Applications (Table [2ddesktopviz])
3D Desktop Applications
Web-Based Visualization
QSAR/ADMET Modeling
Descriptor Calculators
Model Building
Model Application
Visualization
Quantum Chemistry
Ab initio Calcuation
Helper Applications
Visualization
Ligand Dynamics and Free Energy Calculations
Simulation Software
Simulation Setup and Analysis
Virtual Screening and Ligand Design
Ligand-Based
Docking and Scoring
Pocket Detection
Ligand Design
Added to Comp Chem Page
Web browsers used in Drug Discovery
Last week I posted this observation
More and more of the companies/groups that I'm working with are moving away from desktop applications to providing a web-based portfolio of applications for drug discovery. Most seem to use a combination of commercial tools with a selection of in house apps. Whilst this has many advantages it does raise the question about which web browser should they support? Whilst NetMarketshare still has Internet Explorer at 44% this is probably not a good metric to measure browser usage in the Drug Discovery Sector. So for the last couple of months I've been monitoring the web browsers used to access the Drug Discovery Resources since it is unlikely that anyone not interested in drug discovery would spend much time browsing these pages. The results are interesting.
The ranking since 1 Jan 2016 to date is
- Chrome 55%
- Safari 20%
- Firefox 16%
- Internet Explorer 4%
Looking at operating systems
- Windows 57%
- Macintosh 23%
- iOS 11%
- Android 8%
So the lack users of Internet Explorer is not due to the absence of Windows users. This must have implications for all developers, the users appeared to have moved to the more modern web browsers.
Update
I've now data from around 10 different sites involved in drug discovery or software/databases to support drug discovery, ranging from small sites with about 10,000 hits a month to major sites with many millions of hits a month, and I've now included the average data in the table below.
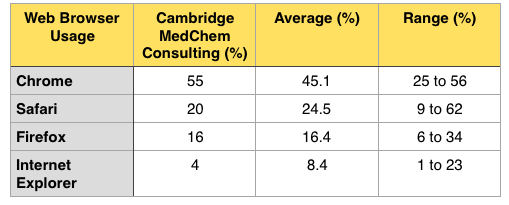
It looks like the data from Drug Discovery Resources reasonably reflects the usage in the Drug Discovery sector.
Web-based tools
More and more of the companies/groups that I'm working with are moving away from desktop applications to providing a web-based portfolio of applications for drug discovery. Most seem to use a combination of commercial tools with a selection of in house apps. Whilst this has many advantages it does raise the question about which web browser should they support? Whilst NetMarketshare still has Internet Explorer at 44% this is probably not a good metric to measure browser usage in the Drug Discovery Sector.
So for the last couple of months I've been monitoring the web browsers used to access the Drug Discovery Resources since it is unlikely that anyone not interested in drug discovery would spend much time browsing these pages. The results are interesting.
The ranking since 1 Jan 2016 to date is
- Chrome 56%
- Safari 20%
- Firefox 16%
- Internet Explorer 4%
Looking at operating systems
- Windows 57%
- Macintosh 23%
- iOS 11%
- Android 8%
So the lack users of Internet Explorer is not due to the absence of Windows users. This must have implications for all developers, the users appeared to have moved to the more modern web browsers.
Update
A number of readers/companies have contacted me since I published with broadly similar results, I hope to compile and publish the anonymised results next week.
Worth a look.
The a third edition of the popular book, The Organic Chemistry of Drug Design and Drug Action by Silverman and Holladay has just been released, I’ve added it to the book list.
Vortex users might be interested in a new script that implements an interesting paper from Wagner et al Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties DOI that describes an algorithm to score compounds with respect to CNS penetration.
Lilly MedChem rules can now be installed using Homebrew. In late 2012 Robert Bruns and Ian Watson published a paper entitled Rules for Identifying Potentially Reactive or Promiscuous Compounds DOI. This article describes a set of 275 rules, developed over an 18-year period, used to identify compounds that may interfere with biological assays, allowing their removal from screening sets.