
Predicting sites of metabolism
I updated the page on predicting metabolism
https://www.cambridgemedchemconsulting.com/resources/ADME/predicting_metabolism.html.
Updated Drug Discovery Resources
Updated the page on metabolism https://www.cambridgemedchemconsulting.com/resources/ADME/metabolism.html.
And the page on covalent ligands https://www.cambridgemedchemconsulting.com/resources/lead_identification/covalent.html.
CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes
CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes DOI, https://nerdd.univie.ac.at/cyplebrity/, structures can be submitted in SMILES format or drawn using the sketcher. The calculation takes a few second per compound and the results are displayed as shown below.
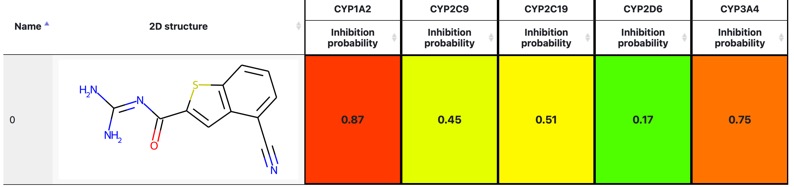
Added to the section on CYP Interactions.
Predicting sites of metabolism
I've updated the predicting sites of metabolism page
https://www.cambridgemedchemconsulting.com/resources/ADME/predicting_metabolism.html
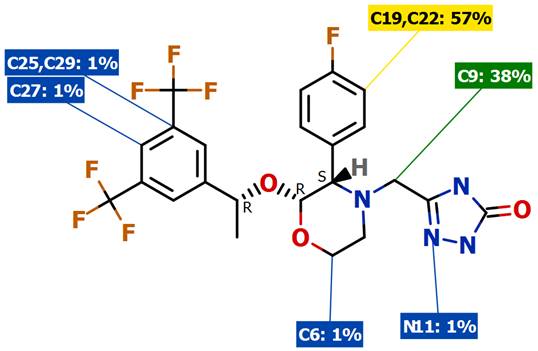
Predicting sites of metabolism
I've added GLORYx to the predicting metabolism page.
GLORYx predicts phase I and phase II metabolites for the chemical compound(s) provided by the user. The method is based on the FAME site of metabolism (SoM) prediction combined with sets of reaction rules encoding both phase I and phase II metabolic reactions.
I tried a range of other molecules and GLORYx was really very impressive in identifying potential metabolites.
Potential 2019-nCoV 3C-like Protease Inhibitors
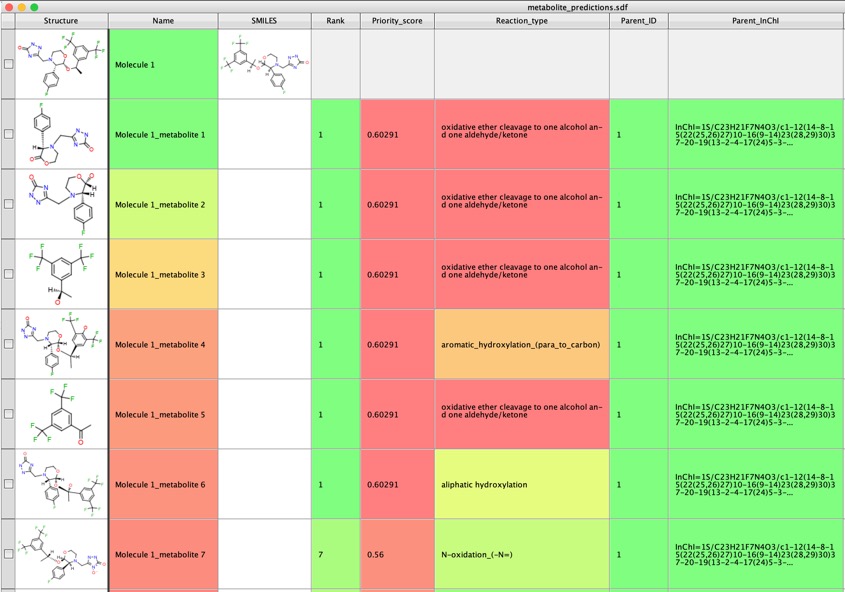
Chris Southan recently flagged a number of publications describing possible treatments for 2019-nCoV using repurposed existing drugs "Therapeutic options for the 2019 novel coronavirus (2019-nCoV)" DOI. In addition a recent preprint "Potential 2019-nCoV 3C-like Protease Inhibitors Designed Using Generative Deep Learning Approaches" DOI highlighted the design of potential protease inhibitors. The authors provide the structures of the molecules in the supplementary informations.
I downloaded the sdf file and used a Jupyter notebook to calculate a range of physicochemical properties, the results are shown in the plot below.
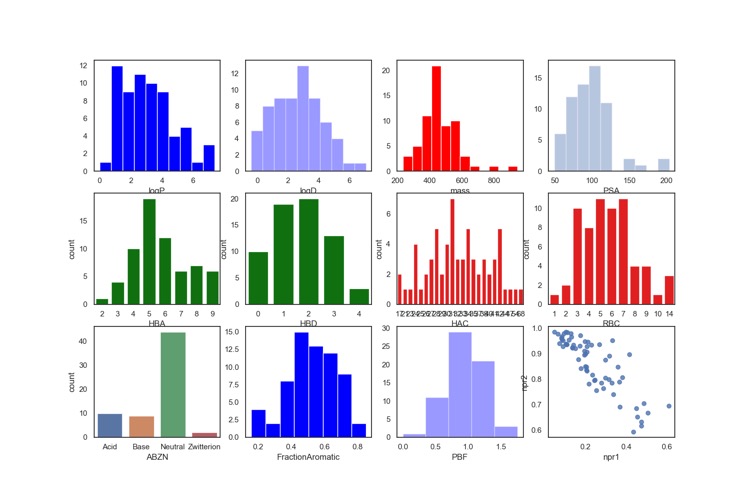
As often seen with protease inhibitors, the molecular weight is rather high with the majority of compounds having Mol Weight >500. The calculated LogP is mainly in the range 2 to 5, however because 40% of the molecules are predicted to be basic the LogD is mainly in the range 0-4. This combination of high molecular weight and rather high LogP is likely to compromise the developability score (for more details on develop ability score read "20 years Rule of Five" report here.
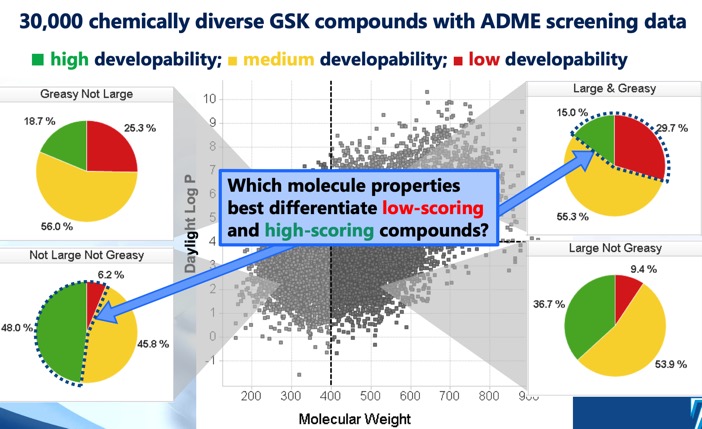
The molecules are also predicted to have a rather high number of hydrogen bond donors and acceptors, this contributes to high polar surface area (TPSA). In general TPSA >120 are often associated with poor oral bioavailability, however it should be noted that the TPSA was not calculated on a 3D structure and it is possible that intramolecular hydrogen bonds may reduce the actual TPSA, also described in the 20 years Rule of Five report.
Scanning through the molecules I noticed a number of functional groups that might be a concern (e.g. Micheal Acceptors), I ran a couple of Vortex scripts that flag potential problematic groups based on SMARTS taken from the following publications
http://pubs.acs.org/doi/abs/10.1021/jm901137j
http://pubs.acs.org/doi/abs/10.1021/jm5019093
https://doi.org/10.1177/1087057116639992
https://dx.doi.org/10.1177%2F1087057113516861
I also ran a couple scripts that flag potential liver toxicity or HERG liabilities. These flags should not be used to exclude molecules but should be used to flag molecules for checking experimentally. The script identifies the functional group that has been flagged as a liver toxicity liability, and identifies the most similar molecule in ChEMBL that has HERG activity. The results are shown in the image below.
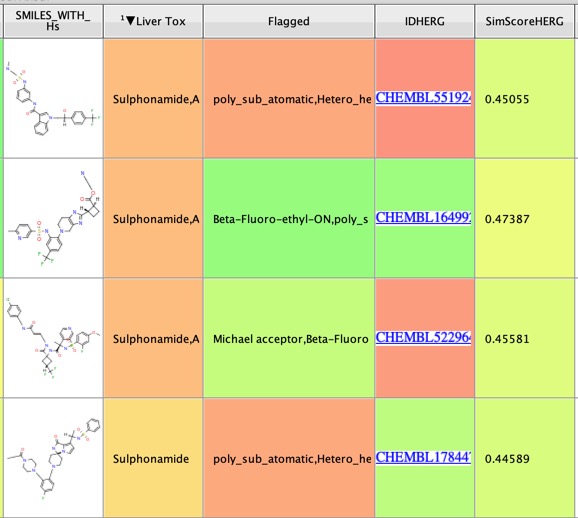
I also added InChiKeys for better cross referencing.
I've exported all results as an sdf file which can be found here http://cambridgemedchemconsulting.com/news/files/INSCoV_2020sdfv1addedflags.sdf.zip.
ADME pages updated
I've spent a while updating the ADME section of the Drug Discovery Resources. I particular I've added a little on the Developability score DOI that identifies four distinct cLog P/molecular weight regions that define optimal and sub-optimal chemical space. I've also added a couple of useful references.
In addition, I've expanded the Absorption and Bioavailability page to include more on bioavailability with links to physicochemical properties. The Distribution and Plasma Protein Binding section has a couple of extra examples demonstrating the impact plasma protein binding has on other pharmacokinetic properties. I've added a few details of in vitro assays to the Transporters page, and expanded the in silico brain penetration models section.
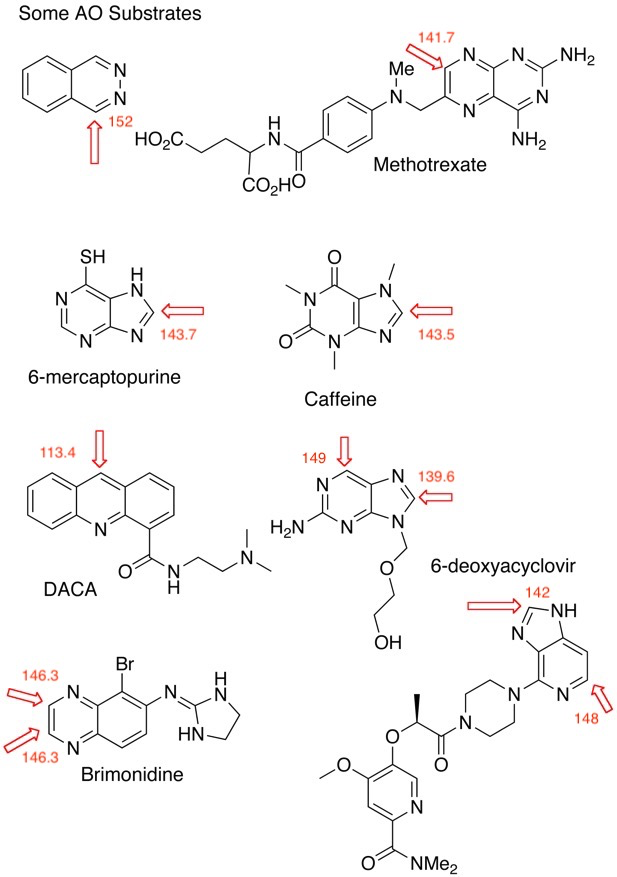
The section on Aldehyde oxidase has been greatly expanded and now includes a section on prediction and mitigation, and added useful references.
Updated Drug Discovery Resources
I've done some updates to the Drug Discovery Resources.
The Following Pages have been Updated
Macrocycles
Predicting Metabolism
Covalent Inhibitors
PROteolysis TArgeting Chimeras (PROTACs), Lysosome Targeting Chimeras (LYTACs), and ENDosome TArgeting Chimeras (ENDTACs)
Drug Discovery Resources Updated
I've spent a little time updating the Drug Discovery Resources Section of the website. In particular:
- CYP interactions now includes details of published crystal structures and more information on known inhibitors
- I've added page on CYP1A2 from ChEMBL data and updated the CYP2D6 and CYP3A4 pages
- Updated the page on Aldehyde Oxidase
- Added new examples on the bioisosteres page
- Updated the Published Fragments section, including adding the overlay of all examples of Kinase fragment hits from the PDB.
- Added new examples to the Chemical Probes page
- Included more examples of halogen bonding to the Molecular Interactions page
Drug Discovery Resources Updated
I've updated the Bioisosteres section adding a few more examples of aryl ring bioisosteres, and I've added CypReact to the predicting metabolism page.
Drug Discovery Resources Updated
I've made a few additions and updated to the Drug Discovery Resources pages. In particular I've updated the covalent inhibitors page and added additional examples to the molecular interactions page. I've also started updating the ADME section and added a page on half-life and how it might be modulated.
Updated brain penetration page
I've updated the brain penetration page to include data from a recent publication, Small Molecule Kinase Inhibitors for the Treatment of Brain Cancer DOI which discusses the issues with targeting brain and central nervous system cancers.
Predicting Sites of Metabolism page updated
I've updated the Predicting sites of metabolism page.
Solubility
I’ve just updated the page on solubility and added a couple of useful assay references.
Solubility may also have an impact on preclinical assays, limited solubility in preclinical ADMET assays may give a false impression of the compounds profile in in vitro assays. Many of the false positives seen in Fragment-based screening are thought to be due to poor solubility at the high concentrations used in the screen. Perhaps the most important is the impact poor solubility can have on gastrointestinal absorption it may also preclude other routes of administration (intravenous).
Using 3Dmol.js
The Drug Discovery Resources pages are intended to act as a resource for scientists undertaking drug discovery, they were initially based on a course I give but have been expanded to give much more detail and to cover subjects not covered in the course. The other advantage of an online resource is that I can include features not possible in static pages.
I had started to include interactive structures a while ago but the problems with java applets and plugins meant I had to abandon that effort. The recent advances in javascript viewers has opened up new possibilities and I've started to reinvestigate more interactive viewers. The initial work uses the fantastic molecule viewer 3Dmol.js developed by David Koes.
3Dmol.js is a modern, object-oriented JavaScript library that uses the latest web technologies to provide interactive, hardware-accelerated three-dimensional representations of molecular data without the need to install browser plugins or Java. 3Dmol.js provides a full featured API for developers as well as a straightforward declarative interface that lets users easily share and embed molecular data in websites.
You can read more about it here DOI.
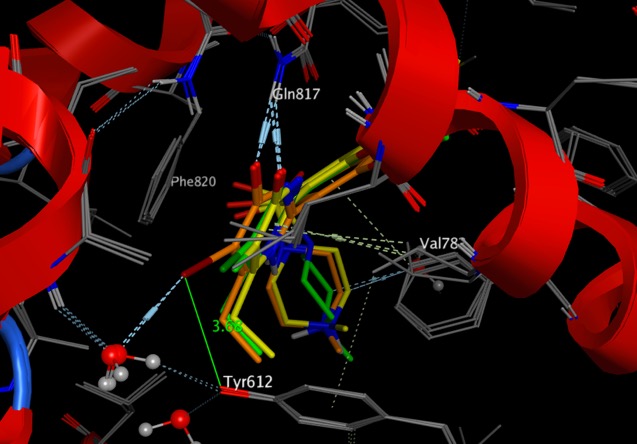
The first page to include an interactive structure is Aldehyde Oxidase, the PDB structure 4UHW is interesting because it shows the binding of both a substrate and an inhibitor binding at a site remote from the active site.
I hope you find this useful and please feel free to contact me with comments and/or suggestions.
Predicting sites of metabolism
I have updated the drug discovery resources on predicting sites of metabolism, I've added several new tools and web-based resources.
Aldehyde Oxidase
I have updated the drug discovery resources page on Aldehyde Oxidase. In particular I have included more detail on the species differences and added the recent X-ray structure of AOX1 with substrate and inhibitor bound.
Insects in Drug Discovery
The company N2MO offers the use of insects as model organisms. They can be used for ADME screening in particular brain penetration studies.
The Grasshopper: A Novel Model for Assessing Vertebrate Brain UptakeOlga Andersson, Steen Honoré Hansen, Karin Hellman, Line Rørbæk Olsen, Gunnar Andersson, Lassina Badolo, Niels Svenstrup, and Peter Aadal Nielsen EntomoPharm R&D, Medicon Village, Lund, Sweden (O.A., K.H., G.A., P.A.N.); Department of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark (S.H.H., L.R.O.); and Division of Discovery Chemistry and Drug Metabolism and Pharmacokinetics, H. Lundbeck A/S, Copenhagen, Denmark (L.B., N.S.) Received April 10, 2013; accepted May 10, 2013
ABSTRACT The aim of the present study was to develop a blood-brain barrier (BBB) permeability model that is applicable in the drug discovery phase. The BBB ensures proper neural function, but it restricts many drugs from entering the brain, and this complicates the development of new drugs against central nervous system diseases. Many in vitro models have been developed to predict BBB permeability, but the permeability characteristics of the human BBB are notoriously complex and hard to predict.
Consequently, one single suitable BBB permeability screening model, which is generally applicable in the early drug discovery phase, does not yet exist. A new refined ex vivo insect-based BBB screening model that uses an intact, viable whole brain under controlled in vitro-like exposure conditions is presented.
This model uses intact brains from desert locusts, which are placed in a well containing the compound solubilized in an insect buffer. After a limited time, the brain is removed and the compound concentration in the brain is measured by conventional liquid chromatography-mass spectrometry. The data presented here include 25 known drugs, and the data show that the ex vivo insect model can be used to measure the brain uptake over the hemolymph-brain barrier of drugs and that the brain uptake shows linear correlation with in situ perfusion data obtainedinvertebrates.Moreover,this study shows that the insect ex vivo model is able to identify P-glycoprotein (Pgp) substrates, and the model allows differentiation between low-permeability compounds and compounds that are Pgp substrates.
Metrabase
p>The Metabolism and Transport Database (Metrabase) is a cheminformatics and bioinformatics resource that contains curated data related to human small molecule metabolism and transport, Journal of Cheminformatics 2015, 7:31 DOI. Currently it includes interaction data on 20 transporters, 3438 molecules and 11649 interaction records manually abstracted from 1211 literature references and supplemented with data from other resources as shown in the image below taken from the original publication.
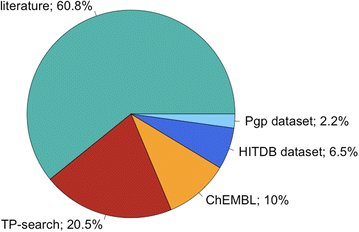
I've added this and more details to the Transporters page of the Drug Discovery Resources
CYP Interactions
Prediction of Cytochromes P450 Inhibition, Bioinformatics, 2013, 29, 2051-2052 WhichCyp, a tool for prediction of which cytochromes P450 isoforms (among 1A2, 2C9, 2C19, 2D6 and 3A4) a given molecule is likely to inhibit. The models are built from experimental high-throughput data using support vector machines and molecular signatures.
Drug Discovery Resources Updates
I’ve added two new pages to the ADME section, there are now separate pages for CYP2D6 inhibitors and CY3A4 inhibitors.
22 July 2014 Updated to include CYP2C9 and CYP2C19 inhibitors.
Aldehyde Oxidase page updated
I’ve updated the page on Aldehyde Oxidase, an enzyme in metabolism of a wide variety of nitrogen heterocycles.
I’ve also included A recent publication DOI that suggests a simple test for the early identification of heteroaromatic drug candidates that have a high probability of metabolism by AO. Bis(((difluoromethyl)sulfinyl)oxy)zinc (DFMS) was used as a source of the CF2H racial, simple LCMS was then used to identify a characteristic M+50 peak. It is also possible to scale up and isolate these metabolically blocked compounds and retest them for improved qualities.
A review of FAst MEtabolizer (FAME)
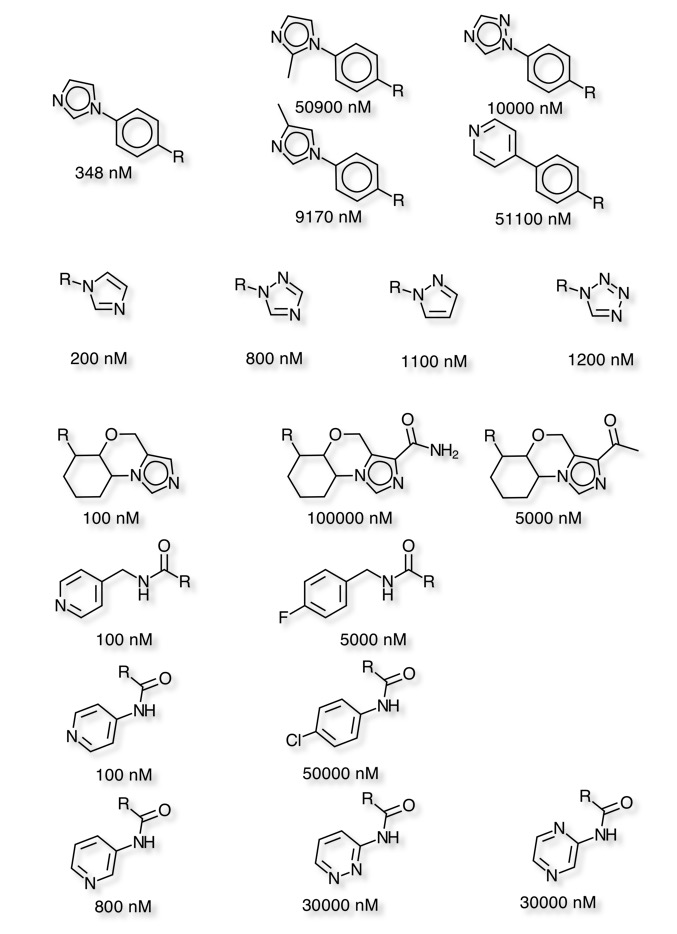
Whilst much computational work is undertaken to support, library design, virtual screening, hit selection and affinity optimisation the reality is that the most challenging issues to resolve in drug discovery often revolve around absorption, distribution, metabolism and excretion (ADME). Whilst we can measure the levels of parent drug in various medium tracking metabolic fate can often be a considerably more difficult proposition requiring significant resources. For this reason prediction of sites of metabolism has become the subject of current interest.
FAME DOI is a collection of random forest models trained on a comprehensive and highly diverse data set of 20,000 small molecules annotated with their experimentally determined sites of metabolism taken from multiple species (rat, dog and human). In addition dedicated models are available to predict sites of metabolism of phase I and II processes.
FAME offers a high performance prediction of sites of metabolism mediated by a wide variety of mechanisms.
Plasma Protein Binding
I’ve just updated the section on distribution and plasma protein binding in the Drug Discovery Resources.
Suggested Books
I’ve just updated the list of suggested books.
Included books on bioisosteres and fragment-based screening.
SMARTCyp 2.4 released
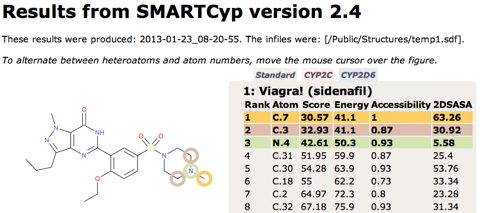
Drug Discovery Resources Update
I’ve updated the Drug Discovery Resources Pages over the Christmas Break. In particular I’ve updated the Fragment Based Screening section and added a page on building a fragment collection. I’ve also updated the section on CYP interactions, expanding the Induction section.
SMARTCyp Updated
SMARTCyp 2.3 has been released with some additional improvements including: Improved energies for N-oxidations Empirical correction for unlikely N-oxidations of tertiary alkylamines A filtering functionality for excluding compounds with very low activation barriers to CYP-mediated oxidations A smiles string can now be input directly on the command line using the -smiles flag. Available as usual at http://www.farma.ku.dk/smartcyp The science behind the improved N-oxidations and the empirical correction has also been published in a paper in Angewandte Chemie: DOI
CYP450 Induction
Updated Aldehyde Oxidase page
I’ve updated the page on Aldehyde Oxidase (AOX1), an enzyme involved in the metabolism of many nitrogen containing heterocycles.
Aldehyde Oxidase
I’ve added a section on Aldehyde Oxidase (AOX1), an enzyme involved in the metabolism of many nitrogen containing heterocycles.
Updated Reading List
Cyprotex ADME Guide: An excellent guide to understanding and interpreting ADME assays and results, available on request from the Cyprotex home page.