
The PROTACtable genome
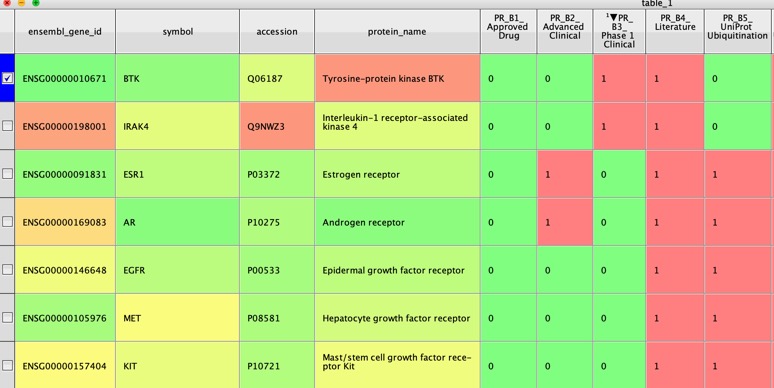
As PROTACs have become more widespread the obvious question is which proteins are best suited to modulation by Protacs? A recent publication provides useful guidelines The PROTACtable genome DOI. The workflow is based on a method developed by a group at GSK, subsequently expanded and now integrated into the Open Targets Platform. Using publicly available data sources, the new method assesses whether a protein could be targeted using a PROTAC, based on the protein’s sequence, location, natural turnover rate in the cell, and evidence from published literature. The framework will help drug discovery researchers to gauge the PROTACtability of their protein of interest, and to prioritise their research accordingly.
More details on PROTACs here
The Chemical Probes Portal now includes several PROTACs
The Chemical Probes Portal now includes several PROTACs in the list of recommended chemical probes and even more are under review by the Scientific Advisory Board.
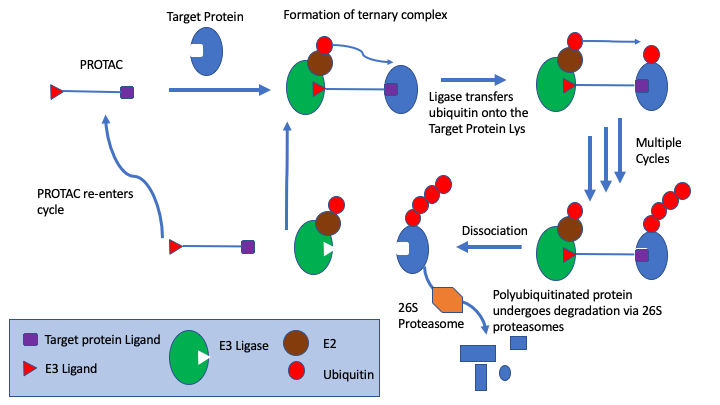
PROTACs are bifunctional molecules that bind to the target protein and an E3 ligase, the simultaneous PROTAC binding of two proteins brings the target protein in close enough proximity for polyubiquitination by the E2 enzyme associated to the E3 ligase, which flags the target protein for degradation through the proteasome.
They have also put together a really useful list of criteria to help define the best choice of Protac probe.
Lysosome Targeting Chimeras (LYTACs)
A while back I added a page on PROteolysis Targeting Chimera (PROTAC) a technology using the ubiquitin proteasome system to induce degradation of the target protein DOI. However this technology is limited to cytosolic proteins.
A recent publication highlights a new technology "Lysosome Targeting Chimeras (LYTACs) for the Degradation of Secreted and Membrane Proteins" DOI that further extends the protein degradation options.
Targeted protein degradation is a powerful strategy to address the canonically undruggable proteome. However, current technologies are limited to targets with cytosolically-accessible and ligandable domains. Here, we designed and synthesized conjugates capable of binding both a cell surface lysosome targeting receptor and the extracellular domain of a target protein…. LYTACs represent a modular strategy for directing secreted and membrane proteins for degradation in the context of both basic research and therapy.
Website Update
I've spent some time over the last couple of weeks updating and adding new content to the Drug Discovery Resources section of the website.
In particular, s drug targets become more challenging medicinal chemists are looking at alternatives to small molecule competitive inhibitors, the section on covalent inhibitors have been expanded and a new page on PROTACs has been added. PROTACs are bifunctional molecules that bind to the target protein and an E3 ligase, the simultaneous PROTAC binding of two proteins brings the target protein in close enough proximity for polyubiquitination by the E2 enzyme associated to the E3 ligase, which flags the target protein for degradation through the proteasome. This offers a powerful alternative to competitive inhibition.
The Probes & Drugs portal has been added to the chemical probes page, this is a public resource joining together focused libraries of bioactive compounds (probes, drugs, specific inhibitor sets etc.) with commercially available screening libraries.
The page describing commercial fragment screening libraries has been updated to include a couple of new additions and flagging some that seem to be unavailable, if I've missed any feel free to let me know.
The section on hERG has been updated with links to new references and details of hERGcentral.
hERGCentral: A Large Database to Store, Retrieve, and Analyze Compound-Human Ether-à-go-go Related Gene Channel Interactions to Facilitate Cardiotoxicity Assessment in Drug Development. The hERGCentral database hergcentral.org is based on experimental data obtained from a primary screen by electrophysiology against more than 300,000 structurally diverse compounds screened at 1 and 10uM.
Unfortunately the database appears to be no longer available. Whilst the supplementary information for the original publication does not contain the structures of the tested compounds it does reference the PubChem substance ID. I used these identifiers to download the structures of the >300,000 records and combined them with the experimental data provided in the Excel tables in the supplementary information. The complete dataset can be downloaded from the hERG page.
Small molecules can potentially bind to a variety of bimolecular targets and whilst counter-screening against a wide variety of targets is feasible it can be rather expensive and probably only realistic for when a compound has been identified as of particular interest. For this reason there is considerable interest in building computational models to predict potential interactions the page on predicting bioactivities has been expanded.
The section on bioisosteres also have a few new examples.