ChEMBL 35 is out
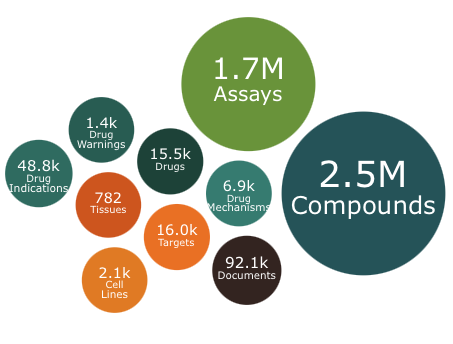
The year ends with an update to ChEMBL. This release contains 2.5 million compounds and 1.7 million assays including over 15K drugs or molecules in development.
You can download the dataset in various formats https://chembl.gitbook.io/chembl-interface-documentation/downloads.
Full details of the update are on the ChEMBL blog. https://chembl.blogspot.com/2024/12/heres-nice-christmas-gift-chembl-35-is.html.
CSD, ChEMBL, PDBe now interlinked
Three critical databases for drug discovery are now interlinked. The Cambridge Structural Database (CSD) a curated repository of small molecule crystal structures, ChEMBL a manually curated database of bioactive molecules with their associated biological data and PDBe a founding member of the Worldwide Protein Data Bank (wwPDB) which collects, organises and disseminates data on biological macromolecular structures, are now interlinked.
The BioChemGraph (BCG) project tackles the challenge of linking diverse data in biology by creating a resource that integrates data from the PDBe, ChEMBL, and the CSD. This has been achieved by mappings UniProt Accession ID and compound InChIKey, linkingmore than 17,000 experimentally determined protein-ligand complexes from the PDB to about 39,000 ChEMBL bioactivity records. By providing this link it is possible to not only identify binding affinity for the selected target but also much more information about the small molecule ligand, such as off-target activities, calculated physicochemical properties and also any ADME/T data that might be available. All data can be downloaded as a tsv as shown below.
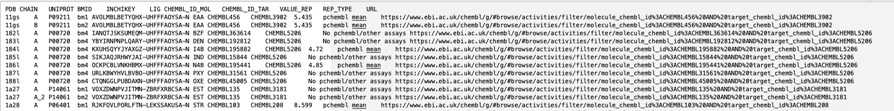
ChEMBL and PDBe have collaborated to set up an automatic pipeline for generating these data. As a result, the data will be updated weekly, in sync with the PDBe release every Wednesday at 00.00 UTC.
InChis have also been used to interconnect with Cambridge Structural Database using UniChem, 235,000 CSD identifiers have been linked corresponding entries in UniChem, a “universal translator” for chemistry using InChIs to connect chemical structures and their identifiers across various databases. UniChem enables researchers to seamlessly access information about a specific molecule across a wide variety of data sources. There are currently 41 data sources (https://www.ebi.ac.uk/unichem/sources).
ChEMBL 33 released
The latest update to ChEMBL has been released.
This fresh release comes with a few new data soures and also some new features: we added bioactivity data for understudied SLC targets from the RESOLUTE project and included a flag for Natural Products and for Chemical Probes. An annotation for the ACTIONTYPE of a measurement was included for approx. 270 K bioactivities. We also time-stamped every document in ChEMBL with their CREATIONDATE!
This version of the database, prepared on 31/05/2023 contains:
2,399,743 compounds (of which 2,372,674 have mol files)
3,051,613 compound records (non-unique compounds)
20,334,684 activities
1,610,596 assays
15,398 targets
88,630 documents
Full details are here http://chembl.blogspot.com/2023/06/release-of-chembl-33.html.
ChEMBL is a manually curated database of bioactive molecules with drug-like properties. It brings together chemical, bioactivity and genomic data to aid the translation of genomic information into effective new drugs.
ChEMBL 32 is released!
The fantastic resource ChEMBL has been updated. ChEMBL 32 contains
- 2,354,965 compounds (of which 2,327,928 have mol files)
- 2,995,433 compound records (non-unique compounds)
- 20,038,828 activities
- 1,536,903 assays
- 15,139 targets
- 86,364 documents
More details are here http://chembl.blogspot.com/2023/03/chembl-32-is-released.html.
Data can be downloaded from the ChEMBL FTP site.
ChEMBL 31 is released
The latest release of the absolutely invaluable ChEMBL database is available.
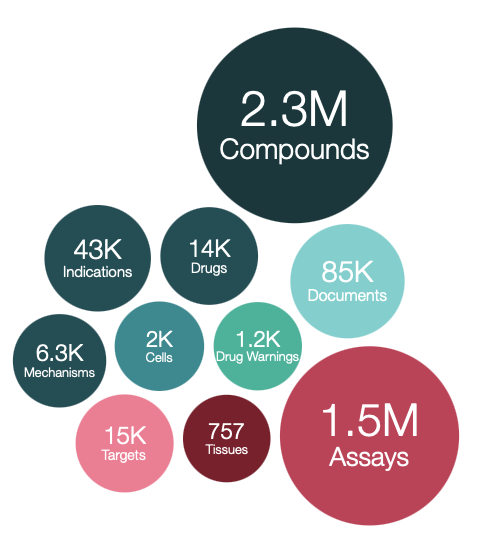
This version of the database, prepared on 12/07/2022 contains:
2,967,627 compound records
2,331,700 compounds (of which 2,304,875 have mol files)
19,780,369 activities
1,498,681 assays
15,072 targets
85,431 documents
Available from the downloads page https://chembl.gitbook.io/chembl-interface-documentation/downloads
ChEMBL 28 has been released.
I see that a new version of ChEMBL has been released. Chembl 28
- 2,680,904 compound records
- 2,086,898 compounds (of which 2,066,376 have mol files)
- 17,276,334 activities
- 1,358,549 assays
- 14,347 targets
- 80,480 documents

ChEMBL 26 Released
The latest release of the essential molecule bioactivity dataset has just been announced.
ChEMBL 26 contains
- 2,425,876 compound records
- 1,950,765 compounds (of which 1,940,733 have mol files)
- 15,996,368 activities
- 1,221,311 assays
- 13,377 targets
- 76,076 documents
A couple of notes
We are now using RDKit for almost all of our compound-related processing. For the first time in ChEMBL26, this will include compound standardization, salt-stripping, generation of canonical smiles, structural alerts, image depiction, substructure searches and similarity searches (via FPSim2: https://github.com/chembl/FPSim2). Therefore, all molecules have been reprocessed and you may notice some differences in molfiles, smiles and structure search results compared with previous releases. The ChEMBL structure curation pipeline has been released as an open source package: https://github.com/chembl/ChEMBLStructure_Pipeline, and incorporated into our Beaker web services (see below). More information can be found here: http://chembl.blogspot.com/2020/02/chembl-compound-curation-pipeline.html.
We are also now using ChemAxon tools to calculate most acidic and basic pKa, logP and logD (pH 7.4) predictions, rather than ACDLabs software. These properties have therefore been recalculated and renamed in the database.
The release notes contain more details and the database can be downloaded from the ChEMBL FTP site.
The Polypharmacology Browser PPB2
Off-target activity is often ignored and might only be uncovered relatively late in the drug discovery program. Whilst broad spectrum screening is available it can be rather expensive. Predicting potential off-target activities is an attractive approach and this paper describes the development of a prediction tool using nearest neighbours combined with machine learning.
The Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning DOI
To build PPB2 we collected a bioactivity dataset of all compounds having at least IC50 < 10 uM on a single protein target in ChEMBL22 considering only high confidence data points as annotated in ChEMBL and only targets for which at least 10 compounds were documented
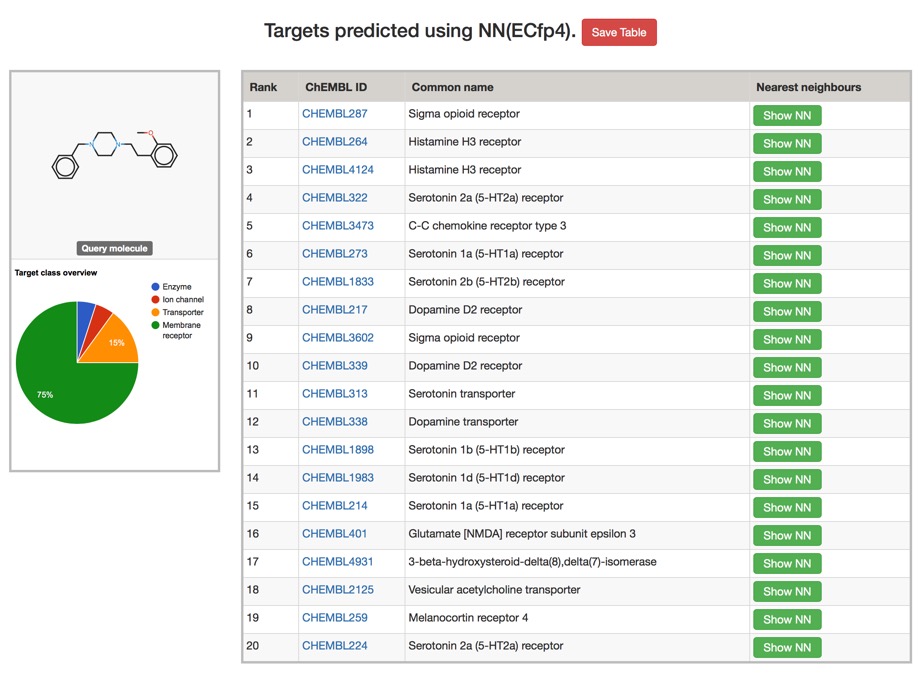
You can try it out here PPB2., depending on the model chosen the results are calculated in a couple of minutes, but don't post your proprietary molecules. Typical results are shown below, clicking on the green "Show NN" button shows the most similar structures.