
Machine Learning for toxicity prediction
Recently there has been an effort to reduced the number of animals used in safety studies for new medicines, and part of that effort has been the increased use of machine learning for toxicity prediction. However, this has proved to be very challenging due to the limited and potentially biased data available.
This open access paper describes strategy for future work DOI
We focus on five crucial pillars for success with ML-driven molecular property and toxicity prediction: (1) data set selection, (2) structural representations, (3) model algorithm, (4) model validation, and (5) translation of predictions to decision-making. Understanding these key pillars will foster collaboration and coordination between ML researchers and toxicologists, which will help to advance drug discovery and development.
3rd In Silico Toxicology Conference
The 3rd In Silico Toxicology Conference, supported by the British Toxicology Society (BTS), the Royal Society of Chemistry (RSC) CICAG Group, Lhasa Ltd., and the Cambridge Alliance on Medicines Safety (CAMS) will take place online on 29 September 2022; attendance is free and open to everyone interested.
Topics will include In Silico Toxicology Consortia, Cell Painting, Gene Expression Data, Biomarkers, Interpreting Neural Networks, Drug-Induced Liver Injury/DILI, Skin Sensitization, Animal Histopathology Data, Species Concordance, In Vivo Pharmacokinetics (PK), Molecular Initiating Events (MIEs), Chemicals, Pharma, Food, Read-Across, ... and beyond (see website for the full programme and registration).
'In Silico Toxicology' Network Meeting 2020
The 'In Silico Toxicology' Network Meeting 2020 will be held on 30 September 2020, 10am-5pm (UK time).
On Zoom this year, and open to all (max 300 participants) more details and registration here http://www.drugdiscovery.net/tox2020/.
An event (with free registration) to foster the In silico Toxicology Community in the UK and beyond. Scientific contributions are welcome as are those on ongoing work, regulatory aspects, industry perspectives, databases, relevant software, etc. in the field. This event is meant to stimulate interactions and discussions, hence speakers are asked to present both about successes and applications that work, as well as areas where still further work is needed, in order to truly develop the field in the future.
In Silico Toxicology' Network Meeting 2019
A new initiative that should be very useful,
An event (with free registration) to foster the In silico Toxicology Community in the UK and beyond. Scientific contributions are as welcome as those on ongoing work, regulatory aspects, industry perspectives, databases, relevant software, journals etc. in the field. This event is meant to stimulate interactions and discussions, hence speakers are asked to present both about in silico toxicology approaches that are already useful to be applied at the current stage, as well as aspects that don't work that well right now, and where future developments are hence needed.
Venue 26 November 2019, Unilever Lecture Theatre, Department of Chemistry, University of Cambridge (CB2 1EW)
Full details and link for registration here In Silico Toxicology' Network Meeting 2019.
hERG central data
A publication by Fang et al DOI describes hERGCentral: A Large Database to Store, Retrieve, and Analyze Compound-Human Ether-à-go-go Related Gene Channel Interactions to Facilitate Cardiotoxicity Assessment in Drug Development. The hERGCentral database hergcentral.org is based on experimental data obtained from a primary screen by electrophysiology against more than 300,000 structurally diverse compounds screened at 1 and 10uM. Unfortunately the database appears to be no longer available. Whilst the supplementary information for the original publication does not contain the structures of the tested compounds it does reference the PubChem substance ID. I used these identifiers to download the structures of the >300,000 records and combined them with the experimental data provided in the Excel tables in the supplementary information. The complete dataset can be downloaded here in either
or in
Bear in mind this is single point data and there will be a fair amount of scatter.
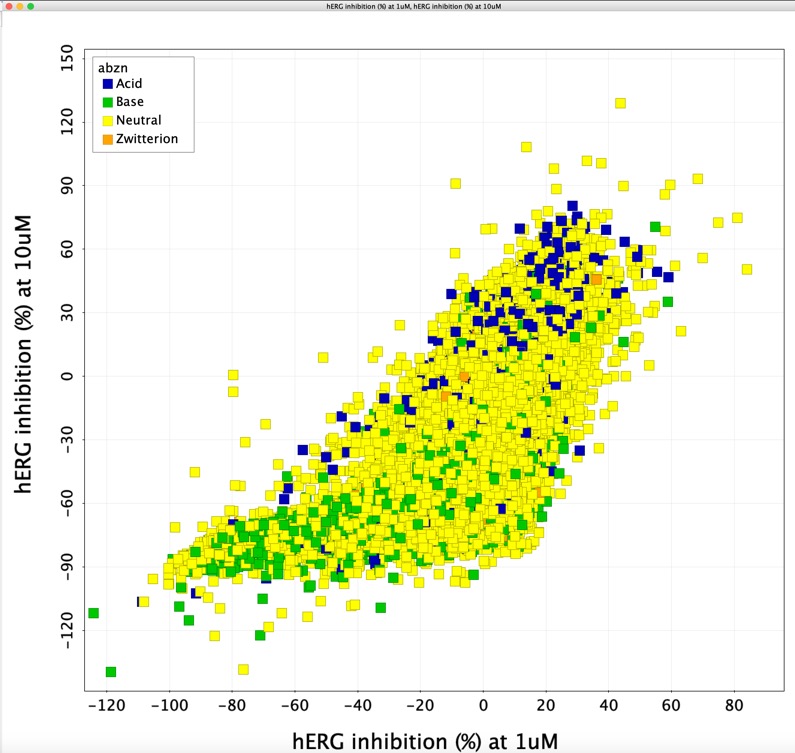
I've added this information to the page on hERG.
Heart on a chip
Cardiotoxicity is a major hurdle for all drug discovery programs and so I'm always interested in ways that any potential liabilities can be flagged without the need to go into whole animals. A recent publication highlights progress in developing a novel model system.
Human induced pluripotent stem cell-derived cardiomyocytes were cultured as a model system, and used to validate the platform with an excitation–contraction decoupling chemical. Preliminary data using the platform to investigate the effect of the drug norepinephrine are combined with computational efforts. This platform provides a quantitative and predictive assay system that can potentially be used for comprehensive assessment of cardiac toxicity earlier in the drug discovery process. DOI.
Hepatotoxicity
I've expended the preclinical toxicity section to include a page on hepatotoxicity. This gives some details of the common assays and markers used to evaluate the potential hepatotoxicity.
Paracetamol Challenge
Sadly it appears that the latest craze to sweep social media is the Paracetamol Challenge in which people (usually children) are encouraged to consume large amounts of the over the counter analgesic paracetamol (called acetaminophen in North America).
One of the particularly insidious features of paracetamol toxicity is that individuals may display little or no symptoms in the first 24h, it is only later when increasing liver damage has occurred do the more serious symptoms become apparent.
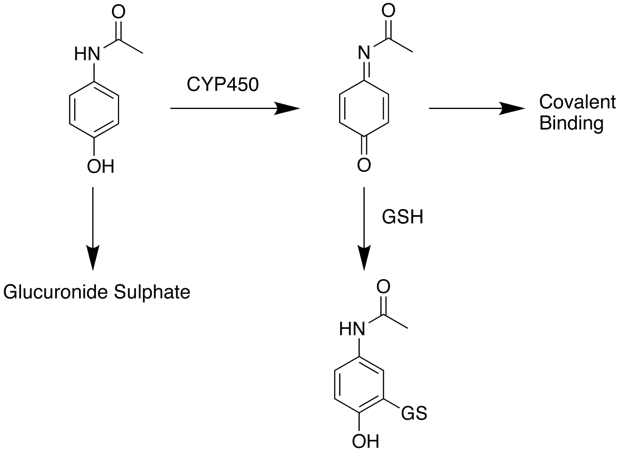
What is the mechanism of Paracetamol Toxicity
At normal therapeutic doses paracetamol the main route for clearance is by Phase II processes including conjugation to form the glucuronide or sulphate followed by renal clearance. At higher doses however, paracetamol is oxidised by CYP450 enzymes in the liver to a highly reactive intermediary metabolite, N-acetyl-p-benzoquinoneimine (NAPQI). NAPQI can be detoxified by conjugation with glutathione (GSH) to form cysteine and mercapturic acid conjugates however this pathway has limited capacity and once supplies of glutathione are exhausted if NAPQI remains it can react covalently with biomolecules resulting in widespread hepatocyte damage and death, leading to acute hepatic necrosis. Without treatment this progresses to irreversible liver, kidney failure followed by multiple organ failures.
Treatment
If caught within an hour of ingestion gastric lavage may be used to remove any drug that has not yet been absorbed, at later stages N-acetylcysteine can be administered. This works to reduce paracetamol toxicity by replenishing body stores of glutathione (GSH). However N-acetylcysteine needs to be administered before liver damage has occurred, if given more than 8 hours after ingestion of paracetamol it's effectiveness is reduced. If patients develop hepatic failure or who are otherwise expected to die from liver failure, the mainstay of treatment is liver transplantation.