
CSD, ChEMBL, PDBe now interlinked
Three critical databases for drug discovery are now interlinked. The Cambridge Structural Database (CSD) a curated repository of small molecule crystal structures, ChEMBL a manually curated database of bioactive molecules with their associated biological data and PDBe a founding member of the Worldwide Protein Data Bank (wwPDB) which collects, organises and disseminates data on biological macromolecular structures, are now interlinked.
The BioChemGraph (BCG) project tackles the challenge of linking diverse data in biology by creating a resource that integrates data from the PDBe, ChEMBL, and the CSD. This has been achieved by mappings UniProt Accession ID and compound InChIKey, linkingmore than 17,000 experimentally determined protein-ligand complexes from the PDB to about 39,000 ChEMBL bioactivity records. By providing this link it is possible to not only identify binding affinity for the selected target but also much more information about the small molecule ligand, such as off-target activities, calculated physicochemical properties and also any ADME/T data that might be available. All data can be downloaded as a tsv as shown below.
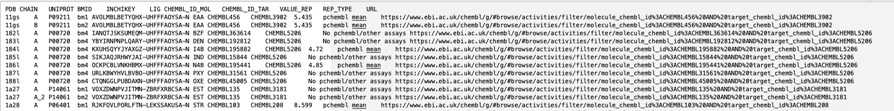
ChEMBL and PDBe have collaborated to set up an automatic pipeline for generating these data. As a result, the data will be updated weekly, in sync with the PDBe release every Wednesday at 00.00 UTC.
InChis have also been used to interconnect with Cambridge Structural Database using UniChem, 235,000 CSD identifiers have been linked corresponding entries in UniChem, a “universal translator” for chemistry using InChIs to connect chemical structures and their identifiers across various databases. UniChem enables researchers to seamlessly access information about a specific molecule across a wide variety of data sources. There are currently 41 data sources (https://www.ebi.ac.uk/unichem/sources).
CCDC: Curated Data Set of Protein Structures
Fantastic news from the Cambridge Crystallographic Data Centre (CCDC), a curated data set of protein structures from the Protein Data Bank (PDB) with predicted hydrogen positions is now available for download. The dataset is taken from the Protein Data Bank (PDB) and has the positions of hydrogens accurately computed, this provides a comprehensive snapshot of protein cavities in the PDB, identifying potential binding sites for small molecules with accurately predicted hydrogen positions for all components.
The news article is here https://www.ccdc.cam.ac.uk/discover/blog/accelerating-drug-discovery-with-the-ccdc-aws-and-intel/.
This large subset of the protein data bank which has be processed using the CCDC's protonation workflow so that reasonable proton positions have been modelled can be downloaded here.
https://www.ccdc.cam.ac.uk/support-and-resources/downloads/.
AlphaFold predicts structure of almost every catalogued protein known to science
A little over a year ago I highlighted the AlphaFold Protein Structure Database in which AlphaFold DB provided open access to protein structure predictions for the human proteome and 20 other key organisms to accelerate scientific research. Well things have moved on.
DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI) have made AI-powered predictions of the three-dimensional structures of nearly all catalogued proteins known to science freely and openly available to the scientific community, via the AlphaFold Protein Structure Database.
The database is being expanded by approximately 200 times, from nearly 1 million protein structures to over 200 million, covering almost every organism on Earth that has had its genome sequenced. The expansion of the database includes predicted structures for a wide range of species, including plants, bacteria, animals, and other organisms.
The full dataset of all predictions is available at no cost and under a CC-BY-4.0 licence from Google Cloud Public Datasets. We've grouped this by single-species for ease of downloading subsets or all of the data. We suggest that you only download the full dataset if you need to process all the data with local computing resources (the size of the dataset is 23 TiB, ~1M tar files).
Downloads can be found here https://alphafold.ebi.ac.uk/download#full-dataset-section.
It is worth noting that AlphaFold2 is not the only protein structure prediction tool available, there is also RoseTTAFold, OpenFold, and FastFold.
AlphaFold Protein Structure Database
The AlphaFold Protein Structure Database Developed by DeepMind and EMBL-EBI is now available online.
AlphaFold DB provides open access to protein structure predictions for the human proteome and 20 other key organisms to accelerate scientific research.
AlphaFold DB currently provides predicted structures for the organisms listed below and includes human, laboratory species, and key pathogens. All the predictions for all the species can be downloaded from the EBI FTP site ftp://ftp.ebi.ac.uk/pub/databases/alphafold.
| Species | Common Name | Reference Proteome | Predicted Structures | Download |
|---|---|---|---|---|
| Arabidopsis thaliana | Arabidopsis | UP000006548 | 27,434 | Download (3642 MB) |
| Caenorhabditis elegans | Nematode worm | UP000001940 | 19,694 | Download (2601 MB) |
| Candida albicans | C. albicans | UP000000559 | 5,974 | Download (965 MB) |
| Danio rerio | Zebrafish | UP000000437 | 24,664 | Download (4141 MB) |
| Dictyostelium discoideum | Dictyostelium | UP000002195 | 12,622 | Download (2150 MB) |
| Drosophila melanogaster | Fruit fly | UP000000803 | 13,458 | Download (2174 MB) |
| Escherichia coli | E. coli | UP000000625 | 4,363 | Download (448 MB) |
| Glycine max | Soybean | UP000008827 | 55,799 | Download (7142 MB) |
| Homo sapiens | Human | UP000005640 | 23,391 | Download (4784 MB) |
| Leishmania infantum | L. infantum | UP000008153 | 7,924 | Download (1481 MB) |
| Methanocaldococcus jannaschii | M. jannaschii | UP000000805 | 1,773 | Download (171 MB) |
| Mus musculus | Mouse | UP000000589 | 21,615 | Download (3547 MB) |
| Mycobacterium tuberculosis | M. tuberculosis | UP000001584 | 3,988 | Download (421 MB) |
| Oryza sativa | Asian rice | UP000059680 | 43,649 | Download (4416 MB) |
| Plasmodium falciparum | P. falciparum | UP000001450 | 5,187 | Download (1132 MB) |
| Rattus norvegicus | Rat | UP000002494 | 21,272 | Download (3404 MB) |
| Saccharomyces cerevisiae | Budding yeast | UP000002311 | 6,040 | Download (960 MB) |
| Schizosaccharomyces pombe | Fission yeast | UP000002485 | 5,128 | Download (776 MB) |
| Staphylococcus aureus | S. aureus | UP000008816 | 2,888 | Download (268 MB) |
| Trypanosoma cruzi | T. cruzi | UP000002296 | 19,036 | Download (2905 MB) |
| Zea mays | Maize | UP000007305 | 39,299 | Download (5014 MB) |
The search bar at the top of the query page accepts queries based on protein name, gene name, UniProt identifier, or organism name. At present you can't search using a sequence and look for similar proteins. You would first need to do a BLAST search and use the results from that as queries.
Here I searched for Plasmodium falciparum carbonic anhydrase (Q8IHW5) a potential Malaria target. As you can see there is no crystal structure in the PDB. Whilst the active site is predicted with high confidence there are clearly regions for which there is very low confidence.

You can then download the structure in PDB or mmCIF format.
I made a homology model (in purple below) of this protein a while back and it has little sequence similarity with any proteins in the PDB. Despite not including a Zinc the Alphafold Predicted Structure includes histidines in positions to potentially coordinate to the Zinc. If it is possible to include the Zinc in the structure prediction I'd be interested in finding out.

Overall I'd say this is a very useful starting point.
How Structural Biologists and the Protein Data Bank Contributed to Recent FDA New Drug Approvals
An interesting review DOI
Discovery and development of 210 new molecular entities (NMEs; new drugs) approved by the US Food and Drug Administration 2010–2016 was facilitated by 3D structural information generated by structural biologists worldwide and distributed on an open-access basis by the PDB. The molecular targets for 94% of these NMEs are known. The PDB archive contains 5,914 structures containing one of the known targets and/or a new drug, providing structural coverage for 88% of the recently approved NMEs across all therapeutic areas. More than half of the 5,914 structures were published and made available by the PDB at no charge, with no restrictions on usage >10 years before drug approval. Citation analyses revealed that these 5,914 PDB structures significantly affected the very large body of publicly funded research reported in publications on the NME targets that motivated biopharmaceutical company investment in discovery and development programs that produced the NMEs.
Macrocycles in the Protein Data Bank
The Protein Data Bank is an absolutely invaluable resource, the PDB is an archive of 3D structural information of proteins, nucleic acids, and bimolecular complex assemblies. However it is much, much more than a simple archive, the submitted structures are curated and annotated to add information about protein ID, sequence information, organism, ligand details etc. This allows users to interrogate the database in many different ways. The database currently holds 141209 entries, with over 10,000 new entries added every year. The vast majority are X-ray crystal structures but there are now over 12,000 NMR derived structures.
The PDB also contains 25626 chemical components - 24007 as free ligands in 106374 PDB file and you can search via a web interface or download the structures in sdf file format. However browsing through the downloaded file it was apparently that macrocycles were not well represented. A discussion with the extraordinarily helpful Rachel Kramer Green at PDB revealed the issue. Basically any ligand containing more that two amino-acids is treated as a protein not a ligand, there are other rules to deal with modified amino-acids etc. but the bottom line is that the only way to get a comprehensive view of macrocycles in the PDB is to download the entire PDB and programmatically by parsing the entire data set.
First we need to decide what size ring constitutes a macrocycle. asking the "internet" failed to produce a definitive answer.
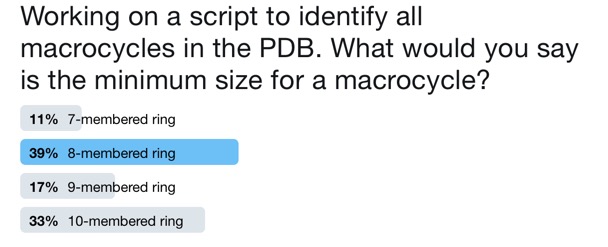
You can read the results and download the script here.
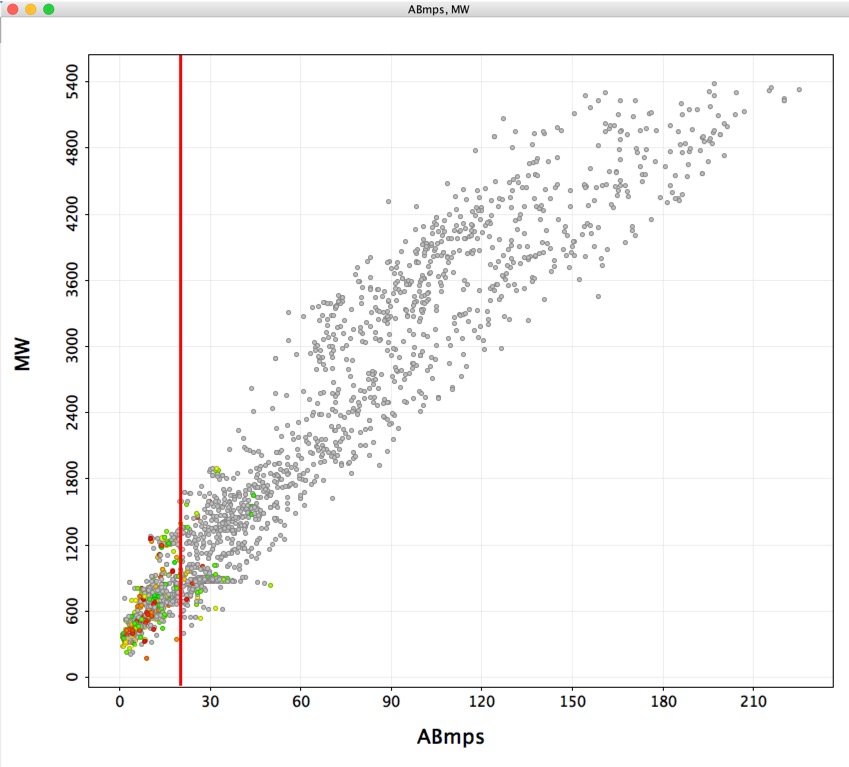
There is also a page that discusses macrocycles in drug discovery.
There is also an upcoming meeting on Macrocycles, 3rd RSC BMCS Medicinal Chemistry Symposium on Macrocycles. Monday-Tuesday, 8th-9th October 2018, GlaxoSmithKline, Stevenage, UK. Full details are here http://www.maggichurchouseevents.co.uk/bmcs/Macrocycles-2018.htm. #BMCS_Macrocycles
The objective of this symposium is to promote scientific interaction between scientists with a shared interest in the field of Macrocycles. This area is responsible for a growing number of therapeutic approaches and development candidates, all of which go ‘beyond the rule-of–five’. As a researcher in this field, come along to hear about the latest advances and also to share in some of the secrets of discovering therapeutic agents which go beyond Lipinski’s rules.