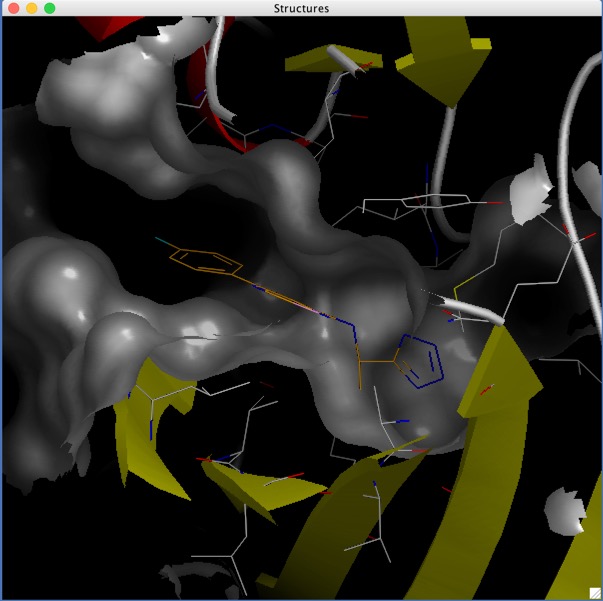
D3R Grand Challenge 2: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies
The Drug Design Data Resource (D3R) is an NIH-funded resource dedicated to improving method development in ligand docking and scoring through community-wide blinded prediction challenges (http://www.drugdesigndata.org). DOI
The Drug Design Data Resource (D3R) ran Grand Challenge 2 (GC2) from September 2016 through February 2017. This challenge was based on a dataset of structures and affinities for the nuclear receptor farnesoid X receptor (FXR), contributed by F. Hoffmann-La Roche. The dataset contained 102 IC50 values, spanning six orders of magnitude, and 36 high-resolution co-crystal structures with representatives of four major ligand classes. Strong global participation was evident, with 49 participants submitting 262 prediction submission packages in total. Procedurally, GC2 mimicked Grand Challenge 2015 (GC2015), with a Stage 1 sub-challenge testing ligand pose prediction methods and ranking and scoring methods, and a Stage 2 sub-challenge testing only ligand ranking and scoring methods after the release of all blinded co-crystal structures. Two smaller curated sets of 18 and 15 ligands were developed to test alchemical free energy methods. This overview summarises all aspects of GC2, including the dataset details, challenge procedures, and participant results. We also consider implications for progress in the field, while highlighting methodological areas that merit continued development. Similar to GC2015, the outcome of GC2 underscores the pressing need for methods development in pose prediction, particularly for ligand scaffolds not currently represented in the Protein Data Bank (http://www.pdb.org), and in affinity ranking and scoring of bound ligands.
Conclusions:
- Successful prediction of ligand–protein poses depends on the entire workflow, including factors extrinsic to the core docking algorithm, such as the conformation of the protein selected.
- The accuracy of pose predictions tends to be improved by the use of available structural data, via ligand overlays and/or selection of receptor structures solved with similar ligands.
- The accuracy of the poses used in structure-based affinity rankings does not clearly correlate with ranking accuracy.
- Explicit solvent free energy methods did not, overall, pro-vide greater accuracy than faster, less detailed scoring methods
Selecting hits from virtual screening
Whilst high-throughput screening (HTS) has been the starting point for many successful drug discovery programs the cost of screening, the accessibility of a large diverse sample collection, or throughput of the primary assay may preclude HTS as a starting point and identification of a smaller selection of compounds with a higher probability of being a hit may be desired. Directed or Virtual screening is a computational technique used in drug discovery research designed to identify potential hits for evaluation in primary assays. It involves the rapid in silico assessment of large libraries of chemical structures in order to identify those structures that most likely to be active against a drug target. The key question is then how many molecules do you select from your virtual screen?
The results of a virtual screening run are effectively a rank ordering of the virtual screening deck ordered by whatever scoring function(s) that have been used. The task then becomes selection of molecules for experimental determination of activity.
I posed this question on the website and the results are shown below. Whilst this obviously a limited snapshot it is interesting that there is a wide variety of responses.
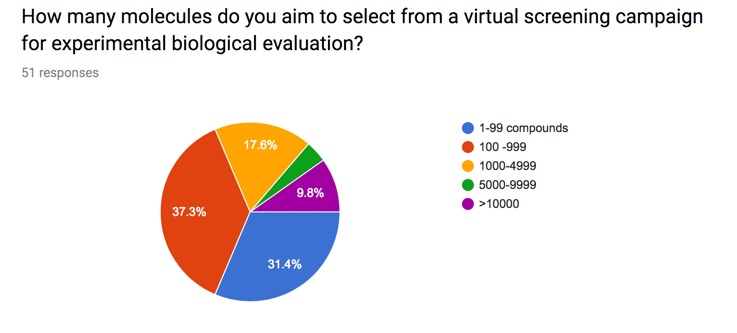
I've included the results on the page on Selecting Compounds from a Virtual Screening Run.
How many compounds do you select from virtual screening?
Whilst high-throughput screening (HTS) has been the starting point for many successful drug discovery programs the cost of screening, the accessibility of a large diverse sample collection, or throughput of the primary assay may preclude HTS as a starting point and identification of a smaller selection of compounds with a higher probability of being a hit may be desired. Directed or Virtual screening is a computational technique used in drug discovery research designed to identify potential hits for evaluation in primary assays. It involves the rapid in silico assessment of large libraries of chemical structures in order to identify those structures that most likely to be active against a drug target. The key question is then how many molecules do you select from your virtual screen?
Whilst virtual screening is certainly less expensive than high-throughput screening it is not free, even an in house academic cluster has an overhead (probably equating to > $10,000 per virtual screen). So with that investment how much would you invest in actual compounds?
Virtual Screening Pages Updated
I've updated the pages describing virtual screening, in particular the docking section.