
Small molecule High Throughput Screen using AstraZeneca facilities
This is a really interesting opportunity. With the demise of the European Lead Factory access to a large, high quality screening deck is very limited.
The MRC now have a unique funding opportunity, a chance to screen against the AstraZeneca screening deck. https://www.ukri.org/opportunity/develop-new-approaches-to-small-molecule-medicine/?utmmedium=email&utmsource=govdelivery.
Funding priority in this round will be given to applications related to fibrosis or extracellular matrix targets.
Two funding opportunities a year with new thematic focus each round. Future areas will include:
- autoimmunity
- pain
- motor neuron disease
- mental health
- dementias (including Parkinson’s and Huntington’s)
- women’s health (including related to metabolic disorders)
Often the challenge for small groups is optimisation of the assay to make it suitable for HTS, so it is great this is also to be funded and AZ will provide technical advice
What will funded, costs related to the staff and consumable costs incurred at AstraZencea for the optimisation and execution of the High Throughput Screen (HTS). These include:
- £20,000 (100% FEC Exceptions) – Optimisation and establishment of an HTS
- £150,000 (100% FEC Exceptions) – Execution of the HTS
- cost of travel, accommodation and subsistence for a host institution researcher to work at AstraZeneca in Cambridge for three months (80% FEC)
- costs for elements of the screening cascade that cannot be undertaken at AstraZeneca and must be undertaken at the host organisation (80% FEC)
- minimal %FTE for the project lead (80% FEC)
Deconstruction of a screening hit
One strategy to investigate screening hits is to simplify the structure to identify the minimum pharmacophore . Whilst lower in affinity the result will have lower molecular weight and LogP. Indeed the molecule may now occupy "fragment space". When I presented this at the recent Fragments meeting a member of the audience coined the phrase "Deconstruction of a screening hit" and several people used the phrase subsequently so perhaps it will used in the future.
Details are here Deconstruction of a screening hit.

Artificial intelligence, engineering biology and quantum technologies: Funding Opportunity
Apply for funding for the application of artificial intelligence (AI), engineering biology, and quantum technologies in biomedical research and development.
You must be based at a UK research organisation eligible for MRC funding.
You can get funding through any grants from MRC responsive mode or translation funding opportunities. You should apply through the existing funding opportunity that is most relevant to your science area and career stage.
We will usually fund up to 80% of your project’s full economic cost.
This highlight notice will be open from 1 April 2024 to 31 March 2025. Applications submitted in this window will be considered for this highlight opportunity. For individual application closing dates refer to the relevant MRC funding opportunity.
9th Fragment-based Drug Discovery Meeting slides
I'm just back from the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ another fabulous meeting and always great to hear about the multitude of ways that Fragments are impacting drug discovery, from target identification, hit discovery to lead optimisation. A number of people asked if the slides from my talk would be available.
Hopefully this link will be accessible to everyone.
http://cambridgemedchemconsulting.com/news/files/FragHitsMar2024.pdf.
9th Fragment-based Drug Discovery Meeting
I'll be heading over to the 9th Fragment-based Drug Discovery Meeting https://www.rscbmcs.org/events/fragments24/ later today. This event is one of the high points in the Drug Discovery calendar. I'm sure there will be plenty to add to the Fragment-Based Screening section on the Drug Discovery Resources Website.
The aim of the 9th RSC-BMCS Fragment-based Drug Discovery meeting will be to continue the focus on case studies in Fragment-based Drug Discovery that have delivered compounds to late stage medicinal chemistry, preclinical or clinical programmes. The Fragment series was started in 2007 and continues with this theme in having over three-quarters of the presentations focused on case studies. This will be complemented by technology progress in high concentration, NMR, SPR and X-ray screening.
BMCS Hot Topics Meetings
The next meeting is Hot Topics: Covalent Drug Discovery 2024, this online event is on Thursday 16th May, 2024 (afternoon).

To register for the meeting, click here To download the first announcement poster, click here
RSC Pharmaceutics has just published its first articles
Pharmaceutics plays a critical role in drug discovery however since it is often regarded as a development process it is not always given the prominence in basic research that it deserves. The pharmaceutical properties of a drug are absolutely critical in the success of a drug discovery project so I'm delighted to see a new open-access RSC journal on the topic
RSC Pharmaceutics has just published its first articles, https://pubs.rsc.org/en/journals/journalissues/pm#!recentarticles&adv.
BBSRC follow-on fund
Grant funding is a great way of starting of work on novel targets, getting funding to continue the work can be more of an issue. This why I'm delighted to read about the BBSRC follow-on fund to help bridge the gap.
FoF applications must draw substantially on current or prior BBSRC funding. You must be based at a UK research organisation eligible for BBSRC funding.
FoF awards aim to take ideas through to a stage where the route to practical application is clear.
The full economic cost (FEC) of your project can be up to £800,000. BBSRC will fund 80% of the FEC. FoF awards support defined programmes of work for up to two years.
Drug design data sets for testing computational tools
One of the challenges when building novel tools to aid drug discovery is identifying high quality datasets that can be used to test new tools. This is why the D3R datasets are so valuable https://drugdesigndata.org.
These datasets are available from BindingDB and include a variety of important protein targets.
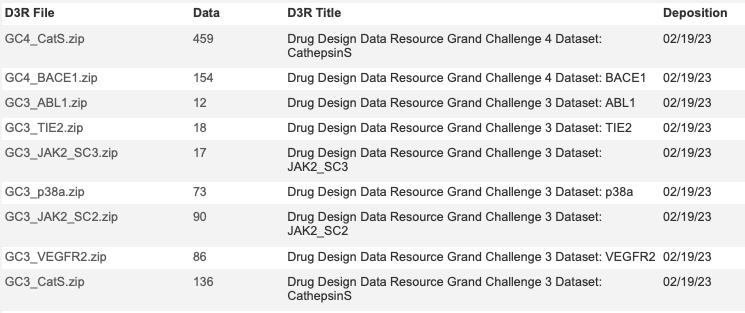
The targets include CathepsinS, BACE1, ABL1) and JAK2.
Seasons Greetings
I hope you all have a great seasonal holiday, it has been a tough few years for folks so I think everyone needs a break. As Bad Company sang in Wishing Well.
But I know what you're wishing for, Love in a peaceful world
As in previous years rather post cards to everyone, instead any monies saved have been donated to the Multiple Sclerosis Society.
