Potential 2019-nCoV 3C-like Protease Inhibitors
Chris Southan recently flagged a number of publications describing possible treatments for 2019-nCoV using repurposed existing drugs "Therapeutic options for the 2019 novel coronavirus (2019-nCoV)" DOI. In addition a recent preprint "Potential 2019-nCoV 3C-like Protease Inhibitors Designed Using Generative Deep Learning Approaches" DOI highlighted the design of potential protease inhibitors. The authors provide the structures of the molecules in the supplementary informations.
I downloaded the sdf file and used a Jupyter notebook to calculate a range of physicochemical properties, the results are shown in the plot below.
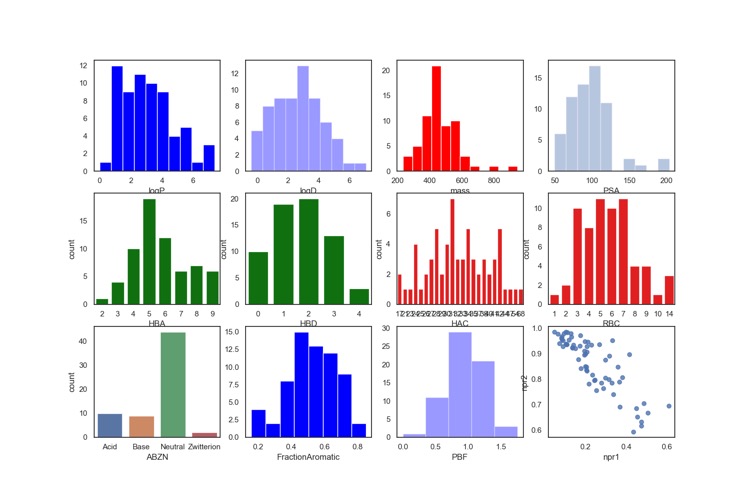
As often seen with protease inhibitors, the molecular weight is rather high with the majority of compounds having Mol Weight >500. The calculated LogP is mainly in the range 2 to 5, however because 40% of the molecules are predicted to be basic the LogD is mainly in the range 0-4. This combination of high molecular weight and rather high LogP is likely to compromise the developability score (for more details on develop ability score read "20 years Rule of Five" report here.
The molecules are also predicted to have a rather high number of hydrogen bond donors and acceptors, this contributes to high polar surface area (TPSA). In general TPSA >120 are often associated with poor oral bioavailability, however it should be noted that the TPSA was not calculated on a 3D structure and it is possible that intramolecular hydrogen bonds may reduce the actual TPSA, also described in the 20 years Rule of Five report.
Scanning through the molecules I noticed a number of functional groups that might be a concern (e.g. Micheal Acceptors), I ran a couple of Vortex scripts that flag potential problematic groups based on SMARTS taken from the following publications
http://pubs.acs.org/doi/abs/10.1021/jm901137j
http://pubs.acs.org/doi/abs/10.1021/jm5019093
https://doi.org/10.1177/1087057116639992
https://dx.doi.org/10.1177%2F1087057113516861
I also ran a couple scripts that flag potential liver toxicity or HERG liabilities. These flags should not be used to exclude molecules but should be used to flag molecules for checking experimentally. The script identifies the functional group that has been flagged as a liver toxicity liability, and identifies the most similar molecule in ChEMBL that has HERG activity. The results are shown in the image below.
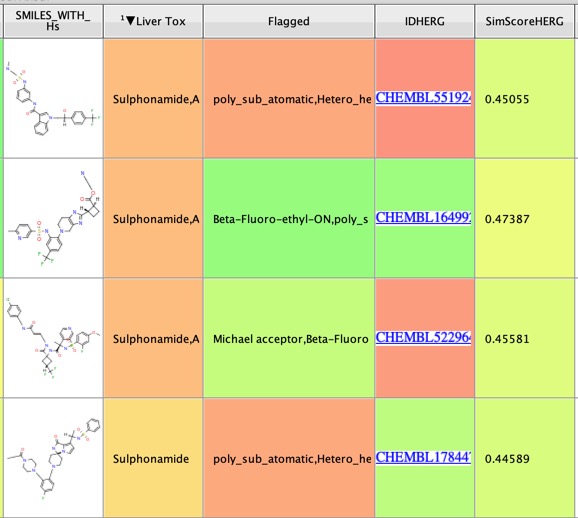
I also added InChiKeys for better cross referencing.
I've exported all results as an sdf file which can be found here http://cambridgemedchemconsulting.com/news/files/INSCoV_2020sdfv1addedflags.sdf.zip.
LogP/D page updated
The page on LogP and LogD is one of the most frequently read and it has been updated to include recent publications.
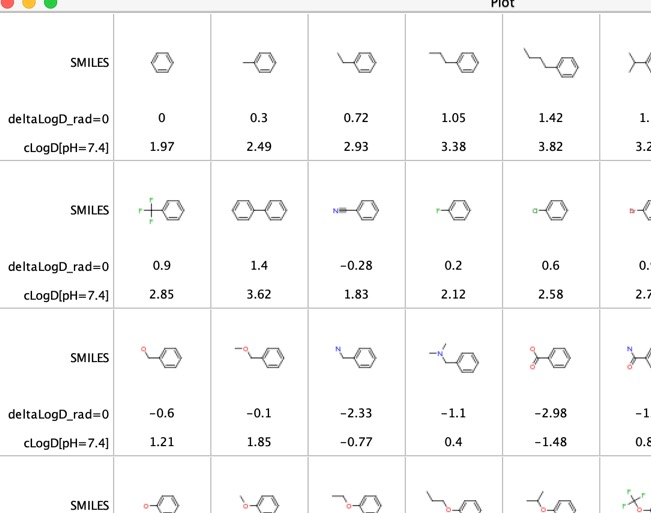
Lipophilicity is possibly the most important physicochemical property of a potential drug, it plays a role in solubility, absorption, membrane penetration, plasma protein binding, distribution, CNS penetration and partitioning into other tissues or organs such as the liver and has an impact on the routes of clearance. It is important in ligand recognition, not only to the target protein but also CYP450 interactions, HERG binding, and PXR mediated enzyme induction.
The contributions of various functional groups to LogD has been explored "LogD contributions of substituents commonly used in medicinal chemistry" DOI, this study used matched molecular pairs analysis of experimental LogD values from several thousand compounds collected using the shake-flask method at pH = 7.4. They reported the average deltaLogD difference for particular molecular pairs. I've compared these experimental results with calculated LogD.