
UKQSAR meeting University of Liverpool on September 14th 2023.
The next UKQSAR meeting will be held at the University of Liverpool on September 14th 2023.
The meeting will take place at building 502, University of Liverpool (see D6 in campus map).
More information will be available on the website https://ukqsar.org/index.php/2023/07/19/uk-qsar-autumn-2023-meeting/
The schedule will be:
9.00-10.00 Registration (coffee/tea/refreshments) 10.00-10.15 Welcome – Nathan Brown (UK QSAR Chair)
10.15-11.45 Session 1 – Chair Neil Berry (University of Liverpool) 10.15-10.45 Talk 1– Alessandro Troisi (University of Liverpool) 10.45-11.15 Talk 2 – Abbie Trewin (University of Lancaster) 11.15-11.45 Talk 3 – Lauren Reid (Medchemica Ltd.) 11.45-12.45 Lunch and poster session
12.45-13.45 Session 2 – Chair Andrew Leach (University of Manchester) 12.45-13.15 Talk 4 – Steve Enoch (Liverpool John Moores University) 13.15-13.45 Talk 5 – Rachel Pirie (University of Newcastle) 13.45-14.15 Break (coffee/tea/refreshments) and poster session
14.15-15.45 Session 3 – Chair Steve Maginn (CCG) 14.15-15.15 Talk 6 – Elena De Orbe (AstraZeneca Ltd.) 15.15-15.45 Talk 7 – Adam Nelson (University of Leeds) 15.45-16.00 Conclusion and poster prizes - Nathan Brown (UK QSAR Chair)
Please register using the online form.
We are delighted that the RSC CICAG group (Chemical Information and Computer Applications Group) have been able to offer two travel bursaries for students to attend the meeting in return for providing a short write-up for the group. If you would like to apply for the bursary please use the form
Ultra large Chemical Libraries
In a recent blog post Derek Lowe talked about "Virtual Screening Versus the Numbers" https://www.science.org/content/blog-post/virtual-screening-versus-numbers highlighting some of the issues around ultra large chemical libraries.
It seems quite timely that RSC CICAG is organising a meeting on Ultra Large Chemical libraries 10 August 2022 10:00-17:00, Burlington House, London, United Kingdom.
A decade ago a chemical library of a million compounds was considered large but over the last few years there has been a period of continuous growth in the size of both physical and virtual chemical libraries. As the libraries have grown the conventional search technologies have become unsustainable and new technologies are needed. This meeting will look at the challenges and solutions used to design, create, compare and search these ultra-large chemical libraries.
There are more details and registration here https://www.rsc.org/events/detail/73675/ultra-large-chemical-libraries.
It is now open for abstract submission (oral due by May 1st, posters June 2nd).
Registration fees
Delegate member early £95
Delegate non-member early £115
Delegate member std £120
Delegate non-member std £145
Student member early £65
Student non-member early £85
Student member std £90
Student non-member std £110
Open-Source Tools workshops
Registration for the next batch of Open-Source Tools workshops run by the RSC Chemical Information and Computer Applications Group is now open.
https://www.eventbrite.com/e/open-source-tools-for-chemistry-tickets-294585512197?.
These workshops have been enormously popular and the interactions with the instructors have been especially valuable. Details of the next 3 workshops are described below.
All meetings start at 2 pm UK time (5 min break after 1 hour). All run using Zoom Webinar
21 April 2022 PDBe Knowledge Base (David Armstrong)
This workshop explores the Protein Data Bank in Europe Knowledge Base (PDBe-KB https://www.ebi.ac.uk/pdbe/) resource and its tools for the investigation, analysis, and interpretation of biomacromolecular structures. PDBe-KB brings together data from all PDB entries and displays this data as aggregated information for individual proteins, including ligand binding sites, macromolecular interactions and more. Furthermore, this community-led resource brings together structural and functional information from a host of other related resources. In this workshop, you will learn how to use the PDBe-KB aggregated views for proteins to investigate structural and function information for proteins and their associated ligands. We will also demonstrate effective use of novel visualisation components of large-scale structural data on these pages, including 3D visualisation of superposed protein structures with their bound ligands.
19 May 2022 KILFS database (Albert Jelke Kooistra, Andrea Volkamer )
Over the past three decades, six thousand structures of the catalytic kinase domain have been made publicly available via the Protein Data Bank. But to what extent are we making use of this wealth of information? In order to harness this data in a better way and to make it readily available for all to use in their research, KLIFS (https://klifs.net) was constructed. KLIFS, i.e. the Kinase–Ligand Interaction Fingerprints and Structures database, is a structural kinase database that systematically collects and processes all structures of the catalytic kinase domain. With the database, you can - for example - easily get a complete overview of all structures, search for ligands with a specific binding mode, identify analogs or your ligands of interest, collect data for your data mining and machine learning applications.
For this workshop, the developers of KLIFS have teamed up with the Volkamer Lab and therefore the workshop will be divided into two segments. First, Albert J. Kooistra will give an introduction to KLIFS and demonstrate different functionalities of the KLIFS website and the integration of KLIFS in KNIME via the 3D-e-Chem nodes. In the second half, Andrea Volkamer and Dominique Sydow will demonstrate, based on their new kinase-focused TeachOpenCADD workflow, how to assess kinase similarity from different data perspectives. They will emphasize their Python package KiSSim – a KLIFS-based kinase structural similarity fingerprint, and OpenCADD-KLIFS – a Python module to facilitate the integration of KLIFS data into kinase research workflows.
23 June 2022 Scoring of shape and ESP similarity (Ester Heid)
Electrostatic effects along with volume restrictions play a major role in enzyme and receptor recognition. Evaluating electrostatic and shape similarities of pairs of molecules such as proposed versus known ligands can therefore be valuable indicators of prospective binding affinities. This workshop will demonstrate how to compute electrostatic and shape similarities using the open-source tool ESP-Sim (github.com/hesther/espsim, doi.org/10.26434/chemrxiv-2021-sqvv9-v3). Available options for comparing electrostatics will be discussed interactively on selected examples of public datasets, along with advice on embedding and aligning molecules prior to computing similarities.
RSC Interest group membership
My RSC membership renewal form has just dropped though the letterbox.

It is the ideal time to think about joining one of the RSC Interest groups.
Interest groups are scientific networks run by members for their community. Each group is themed around a specific area or application of the chemical sciences. They organise an annual series of events to cater for both their members and the wider scientific community. These events vary from: multi-day conferences and workshops to training events.
If you are interested in the computational side of drug discovery why not join the Chemical Information and Computer Applications Group CICAG code 86.
'In Silico Toxicology' Network Meeting 2020
The 'In Silico Toxicology' Network Meeting 2020 will be held on 30 September 2020, 10am-5pm (UK time).
On Zoom this year, and open to all (max 300 participants) more details and registration here http://www.drugdiscovery.net/tox2020/.
An event (with free registration) to foster the In silico Toxicology Community in the UK and beyond. Scientific contributions are welcome as are those on ongoing work, regulatory aspects, industry perspectives, databases, relevant software, etc. in the field. This event is meant to stimulate interactions and discussions, hence speakers are asked to present both about successes and applications that work, as well as areas where still further work is needed, in order to truly develop the field in the future.
SCI-RSC Workshop on Computational Tools for Drug Discovery 2020
The SCI Fine Chemicals Group and RSC Chemical Information and Computer Applications Group are organising a second Workshop on Computational Tools for Drug Discovery. The meeting format will be the same as the very successful meeting run in Birmingham in 2019.
The 2020 workshop will be held on 19 May 2020 at Riverside West, Whitehall Road, Leeds , West Yorkshire, LS1 4AW.
The 2020 workshop will be held on 19 May 2020 at Riverside West, Whitehall Road, Leeds , West Yorkshire, LS1 4AW,
The Workshop Providers and Facilitators are
- Al Dossetter, MedChemica
- Greg Landrum, KNIME
- Gunther Stahl, OpenEye
- Ilenia Giangreco, CCDC
- Matt Segall, Optibrium
- Stuart Firth-Clark, Cresset
Attendees will be able to choose from 4 of 6 sessions.
To select which workshops you would like to attend for each session, please complete the survey on the website. Please note that spaces are allocated on a first-come, first-served basis.
More details of the workshops and registration details are on the website shown below
https://www.soci.org/events/scirsc-workshop-on-computational-tools-for-drug-discovery.
20 Years of Rule of 5 Meeting Report
It has been over twenty years since Lipinski published his work determining the properties of drug molecules associated with good solubility and permeability. Since then, there have been a number of additions and expansions to these “rules”. There has also been keen interest in the application of these guidelines in the drug discovery process and how these apply to new emerging chemical structures such as macrocycles. This symposium brought together researchers from a number of different areas of drug discovery and provided a historical overview of the use of Lipinski’s rules, as well as looking to the future and how we use these rules in the changing drug compound landscape.
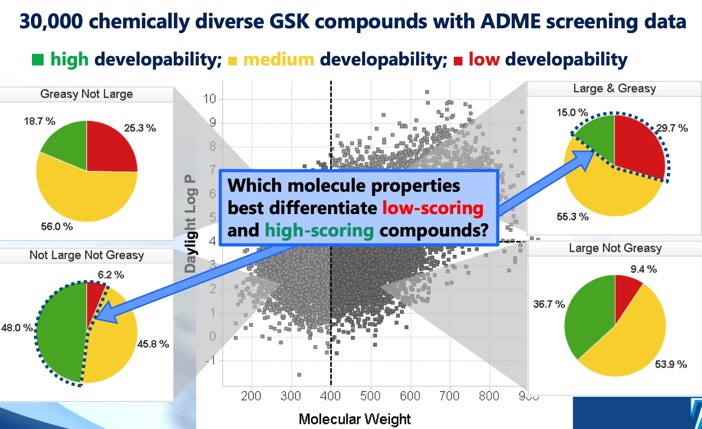
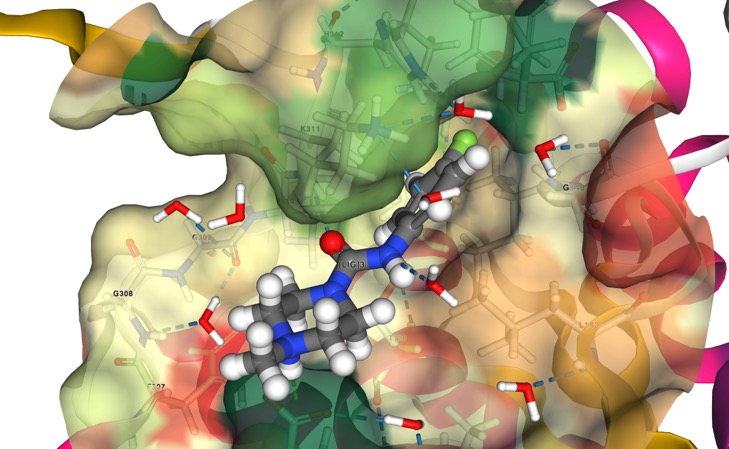
The 20 Years of the Rule of Five Meeting brought together researchers from a number of different areas of drug discovery and provided both a historical overview of the use of Lipinski’s rules, as well as looking to the future and how these rules might evolve in the changing drug compound landscape. The meeting had a capacity attendance of over 100, with Sygnature kindly providing the venue. The audience was a nice mix of industry “veterans”, students and those new to the industry. The meeting format was a morning session giving a historical viewpoint followed by a panel discussion, and the afternoon was dedicated by a more forward looking session again followed by a panel discussion.
The full report is here in PDF format Full Report, many thanks to the presenters for permission to use the images.
More details and the available slide decks are here, Twitter hashtag - #RuleofFive2019.