
Machine Learning for toxicity prediction
Recently there has been an effort to reduced the number of animals used in safety studies for new medicines, and part of that effort has been the increased use of machine learning for toxicity prediction. However, this has proved to be very challenging due to the limited and potentially biased data available.
This open access paper describes strategy for future work DOI
We focus on five crucial pillars for success with ML-driven molecular property and toxicity prediction: (1) data set selection, (2) structural representations, (3) model algorithm, (4) model validation, and (5) translation of predictions to decision-making. Understanding these key pillars will foster collaboration and coordination between ML researchers and toxicologists, which will help to advance drug discovery and development.
Comparison of protein structure prediction algorithms
The majority of drug targets are proteins and knowledge of the 3D structure of the protein can be very helpful for structure based design. Whilst the PDB contains 227,933 structures there are still a number of structures that lack structural information. In 2018 Deepmind released AlphaFold an artificial Intelligence program design to predict protein 3D structure from the amino-acid sequence DOI. Since then there have a series of updates that have included the ability to handle small molecules, co-factors, nucleic acids, protein complexes etc. AlphaFold has been used in collaboration with the EBI to create AlphaFold DB which provides open access to over 200 million protein structures, covering the human proteome and the proteomes of 47 other key organisms important in research and global health. A recent addition is Foldseek a protein structural search program that allows users to search the AlphaFold Database.

David Baker, Demis Hassabis and John Jumper were awarded the 2024 Nobel Prize for Chemistry. One half of the prize has been awarded to David Baker “for computational protein design” and the other half jointly to Demis Hassabis and John M. Jumper “for protein structure prediction.”
Whilst AphaFold gets much of the publicity, it has served to spawn a number of related programs, comparison of the different options is difficult especially when looking at the various licensing options. Fortunately, Brian Naughton has posted a very useful summary. http://blog.booleanbiotech.com/alphafold3-boltz-chai1.html.
AlphaProteo generates novel proteins
Protein protein interactions are always a challenge to optimise and it looks like the latest offering from Google DeepMind may be of significant help.
Protein binders that can bind tightly to a target protein are hard to design. Traditional methods are time intensive, requiring multiple rounds of extensive lab work. After the binders are created, they undergo additional experimental rounds to optimize binding affinity, so they bind tightly enough to be usefu
AlphaProteo generates novel proteins that bind to other proteins. Given the structure of a target molecule and a set of preferred binding locations on that molecule, AlphaProteo generates a candidate protein that binds to the target at those locations.
Whilst code is not available, note
If you’re a biologist, whose research could benefit from target-specific protein binding, and you’d like to register interest in being a trusted tester for AlphaProteo, please reach out to us on alphaproteo@google.com.
LLM for Drug Discovery
Whilst general large language models have hit the headlines in recent years, there is a school of thought that smaller domain specific models may actually more useful, in particular in areas like chemistry https://pubs.rsc.org/en/content/articlelanding/2023/dd/d2dd00087c and https://arxiv.org/abs/2402.09391.
A recent preprint describes Tx-LLM a large language model (LLM) for drug discovery https://arxiv.org/pdf/2406.06316. This work from Google Research and Google DeepMind details Tx-LLM, a LLM specifically designed to enhance drug discovery.
Developing therapeutics is a lengthy and expensive process that requires the satisfaction of many different criteria, and AI models capable of expediting the process would be invaluable. However, the majority of current AI approaches address only a narrowly defined set of tasks, often circumscribed within a particular domain. To bridge this gap, we introduce Tx-LLM, a generalist large language model (LLM) fine-tuned from PaLM-2 which encodes knowledge about diverse therapeutic modalities. Tx-LLM is trained using a collection of 709 datasets that target 66 tasks spanning various stages of the drug discovery pipeline. Using a single set of weights, Tx-LLM simultaneously processes a wide variety of chemical or biological entities (small molecules, proteins, nucleic acids, cell lines, diseases) interleaved with free-text, allowing it to predict a broad range of associated properties, achieving competitive with state-of-the-art (SOTA) performance on 43 out of 66 tasks and exceeding SOTA on 22. Among these, Tx-LLM is particularly powerful and exceeds best-in-class performance on average for tasks combining molecular SMILES representations with text such as cell line names or disease names, likely due to context learned during pretraining. We observe evidence of positive transfer between tasks with diverse drug types (e.g., tasks involving small molecules and tasks involving proteins), and we study the impact of model size, domain finetuning, and prompting strategies on performance. We believe Tx-LLM represents an important step towards LLMs encoding biochemical knowledge and could have a future role as an end-to-end tool across the drug discovery development pipeline.
The model was trained using 709 drug discovery datasets comprising 66 tasks formatted for instruction tuning from Therapeutics instruction Tuning (TxT) https://tdcommons.ai collection for tasks across the drug discovery spectrum. These tasks include:
- Evaluating drug efficacy and safety.
- Predicting molecular targets.
- Assessing the ease of manufacturing drugs.
Artificial intelligence, engineering biology and quantum technologies: Funding Opportunity
Apply for funding for the application of artificial intelligence (AI), engineering biology, and quantum technologies in biomedical research and development.
You must be based at a UK research organisation eligible for MRC funding.
You can get funding through any grants from MRC responsive mode or translation funding opportunities. You should apply through the existing funding opportunity that is most relevant to your science area and career stage.
We will usually fund up to 80% of your project’s full economic cost.
This highlight notice will be open from 1 April 2024 to 31 March 2025. Applications submitted in this window will be considered for this highlight opportunity. For individual application closing dates refer to the relevant MRC funding opportunity.
AlphaFold Protein Structure Database in 2024
A recent publication describes the continued evolution of the AlphaFold Protein Structure Database created by EMBL-EBI and DeepMind. From an initial 300K structures it now contains 214 million predicted protein structures.
You can read the paper here DOI.
The AlphaFold Database Protein Structure Database (AlphaFold DB, https://alphafold.ebi.ac.uk) has significantly impacted structural biology by amassing over 214 million predicted protein structures, expanding from the initial 300k structures released in 2021. Enabled by the groundbreaking AlphaFold2 artificial intelligence (AI) system, the predictions archived in AlphaFold DB have been integrated into primary data resources such as PDB, UniProt, Ensembl, InterPro and MobiDB. Our manuscript details subsequent enhancements in data archiving, covering successive releases encompassing model organisms, global health proteomes, Swiss-Prot integration, and a host of curated protein datasets. We detail the data access mechanisms of AlphaFold DB, from direct file access via FTP to advanced queries using Google Cloud Public Datasets and the programmatic access endpoints of the database. We also discuss the improvements and services added since its initial release, including enhancements to the Predicted Aligned Error viewer, customisation options for the 3D viewer, and improvements in the search engine of AlphaFold DB.
£13 million for 22 AI for health research projects
UKRI have announced £13 invested in medical research projects. The projects aim to transform health using artificial intelligence (AI) to assist and refine diagnostics and procedures
https://www.ukri.org/news/13-million-for-22-ai-for-health-research-projects/
Includes image analysis in oncology, keyhole surgery, NLP analysis of clinical data and treatments for chronic pain.
Fake Publications in Biomedical Science
There have a number of headlines recently highlighting large language models (LLM https://en.wikipedia.org/wiki/Largelanguagemodel, most notably GTP-4 from OpenAI. These models are trained on vast amounts of data from a variety of sources and the quality of these data sources is not always as good as hoped.
It might be assumed the scientific literature would be of a higher standard but a recent preprint raises major concerns.
https://www.medrxiv.org/content/10.1101/2023.05.06.23289563v1
Fake Publications in Biomedical Science: Red-flagging Method Indicates Mass Production
Red-flagged fake publications (RFPs) account for around 28% of the published papers in biomedicine.
AlphaFold predicts structure of almost every catalogued protein known to science
A little over a year ago I highlighted the AlphaFold Protein Structure Database in which AlphaFold DB provided open access to protein structure predictions for the human proteome and 20 other key organisms to accelerate scientific research. Well things have moved on.
DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI) have made AI-powered predictions of the three-dimensional structures of nearly all catalogued proteins known to science freely and openly available to the scientific community, via the AlphaFold Protein Structure Database.
The database is being expanded by approximately 200 times, from nearly 1 million protein structures to over 200 million, covering almost every organism on Earth that has had its genome sequenced. The expansion of the database includes predicted structures for a wide range of species, including plants, bacteria, animals, and other organisms.
The full dataset of all predictions is available at no cost and under a CC-BY-4.0 licence from Google Cloud Public Datasets. We've grouped this by single-species for ease of downloading subsets or all of the data. We suggest that you only download the full dataset if you need to process all the data with local computing resources (the size of the dataset is 23 TiB, ~1M tar files).
Downloads can be found here https://alphafold.ebi.ac.uk/download#full-dataset-section.
It is worth noting that AlphaFold2 is not the only protein structure prediction tool available, there is also RoseTTAFold, OpenFold, and FastFold.
CASP15 details
The details of the latest Critical Assessment of Structure Prediction (CASP) experiment to determine and advance the state of the art in modeling biomolecular structures have been published https://predictioncenter.org/casp15/index.cgi.
Modeling categories
The core of CASP remains the same: blind testing of methods with independent assessment against experiment to establish the state-of-art in modeling proteins and protein complexes. CASP15 will include following categories.
- Single Protein and Domain Modeling As in previous CASPs, the accuracy of single proteins and where appropriate single protein domains will be assessed, using the established metrics. Two changes will be the elimination of the distinction between template-based and template-free modeling, and an emphasis on the fine-grained accuracy of models, such as local main chain motifs and side chains. Because of the high accuracy of the new modeling methods, we expect assessment against high resolution experimental structures will be most informative.
- Assembly As in recent CASPs, the ability of current methods to correctly model domain-domain, subunit-subunit, and protein-protein interactions will be assessed. We will again work in close collaboration with our CAPRI partners. Because of the promising deep learning results reported so far, substantial progress is expected.
- Accuracy Estimation Members of the community will be invited to submit accuracy estimates for multimeric complexes and inter-subunit interfaces. There will no longer be a category for estimating the accuracy of single protein models, since it has become clear these cannot compete with modeling method specific estimates. Instead, there will be increased emphasis on assessment of self-reported accuracy estimates at the atomic level. Note the units will now be pLDDT, not Angstroms.
- RNA structures and complexes There will be a pilot experiment to assess the accuracy of modeling for RNA models and protein-RNA complexes. The assessment will be done in collaboration with the RNA-Puzzles and Marta Szachniuk's group in Poznan.
- Protein-ligand complexes Subject to the availability of adequate resources, there will also be a pilot experiment in this area. Deep-learning is already having an impact here, and there is high interest because of the relevance to drug design.
- Data Assisted As in recent CASPs, there will be assessment of the extent to which the accuracy of models can be increased by the provision of sparse data, particularly that provided by SAXS and mass spectroscopy/chemical crosslinking. Only targets where these low-resolution data are likely to be useful will be considered, that is, large single proteins and complexes. As previously, we will work with collaborators to obtain the necessary experimental data. Targets will initially be released without the experimental data, followed by a second round of prediction including those data.
- Protein conformational ensembles Following the success of deep-learning methods for single structures, it is increasingly important to assess methods for predicting structure ensembles. This is a huge area, ranging from the many conformations of disordered regions to the small number of conformations that may be involved in allosteric transitions and enzyme excited states to local protein dynamics. While it is clear that deep learning and other methods have the potential to generate ensembles in some circumstances, the difficulty is in finding cases where there are sufficiently accurate and extensive experimental data to allow rigorous assessment. One promising avenue is modeling sets of conformations in regions of cryo-EM structures where there is evidence of local conformational heterogeneity. If suitable cases arise, we will present these as a special type of sub-target. First requesting conformational ensembles that will be evaluated against the election density map and then in a possible second stage providing the map for data assisted ensemble prediction. A second possibility is for cases where detailed NMR data have already established the structure of two or more conformations. We have a good lead for a few targets of this type. In addition to this, we are considering a non-blind experiment (a departure from normal CASP practice), where we will first ask those interested to reproduce the known conformations. We will also ask participants to identify any additional conformations that appear to be present. It may then be possible to test these against existing or new experimental data.
Details of the targets will be made available over the next week https://predictioncenter.org/casp15/targetlist.cgi.
AI4Proteins videos now online
On June 16/17 2021 RSC CICAG and AI3D held a joint meeting on Protein Structure Prediction. The full lineup of speakers, titles and abstracts can be found here.
Session 1: Session Chair: Professor Jeremy Frey (University of Southampton)
An AI solution to the protein folding problem: what is it, how did it happen, and some implications Professor John Moult (University of Maryland)
Session 2: Session Chair: Dr Melanie Vollmar (Diamond)
So you predicted a protein structure – What now? Dr Thomas Steinbrecher (Schrödinger)
Deep Learning enhanced prediction of protein structure and dynamics Dr Martina Audagnotto (AstraZeneca)
Fireflies-Lévy Flights algorithm for peptides conformational optimization Dr Zied Hosni (University of Sheffield)
Session 3: Session Chair: Dr Chris Swain (Cambridge MedChem Consulting)
How good are protein structure prediction methods at predicting folding pathways? Mr Carlos Outeiral Rubiera (University of Oxford)
Protein-Ligand Structure Prediction for GPCR Drug Design Dr Chris De Graaf (Sosei Heptares)
Session 4: Session Chair: Dr Márton Vass
Using icospherical input data in machine learning on the protein-binding problem Dr Ella Gale (University of Bristol)
Biological sequence design with machine learning Professor Debora Marks (Harvard University)
Session 5: Session Chair: Dr Simone Fulle (Novo Nordisk)
Lessons learned from generative models of biological sequences Professor Aleksej Zelezniak (Chalmers University of Technology)
DeepDock: a deep learning approach to predict ligand binding conformations Dr Oscar Méndez-Lucio (Janssen Pharmaceuticals)
Finding new in silico-based therapeutic strategies for IAHSP Dr Matteo Rossi Sebastiano (University of Turin)
Session 6: Session Chair: Professor Jonathan Goodman (University of Cambridge)
Designing molecular models by machine learning and experimental data Professor Cecilia Clementi (Freie Universität Berlin)
The “almost druggable” genome Professor Tudor Oprea (University of New Mexico)
Session 7: Session Chair: Dr Lucy Colwell (University of Cambridge)
General Effects of AI on Drug Discovery Dr Derek Lowe (Novartis)
Open Access Data: A Cornerstone for Artificial Intelligence Approaches to Protein Structure Prediction Professor Stephen Burley (RCSB PDB, Rutgers University, UCSD)
The videos of the presentations are now available on YouTube and you can access the playlist here https://www.youtube.com/playlist?list=PLBQwbn0mPhvWyTLnN6eFsbIwb5FByrs.
For those wanting a hype free insight into the impact AI might make on Drug Discovery then the presentation by Derek Lowe is well worth watching.
AI3SD Online Guest Lecture Series
Artificial Intelligence and Augmented Intelligence for Automated Investigations for Scientific Discovery (AI3SD) are running an Online Guest Lecture Series this summer. The full seminar list is here.
http://www.ai3sd.org/summer-seminar-series-2020.
If you missed a presentation or want to replay it, all the presentations are on the AI3SD YouTube channel.
COVID-19 Open Research Dataset Challenge (CORD-19)
There are a number of COVID-19 Kaggle challenges open at the moment, https://www.kaggle.com/datasets?search=COVID.
One of the more recent is:-
COVID-19 Open Research Dataset Challenge (CORD-19)
There is a large body of research and literature continuously evolving around COVID-19. Help the research community and global organizations better digest this to answer key questions."
In response to the COVID-19 pandemic, the White House and a coalition of leading research groups have prepared the COVID-19 Open Research Dataset (CORD-19). CORD-19 is a resource of over 29,000 scholarly articles, including over 13,000 with full text, about COVID-19, SARS-CoV-2, and related coronaviruses. This freely available dataset is provided to the global research community to apply recent advances in natural language processing and other AI techniques to generate new insights in support of the ongoing fight against this infectious disease. There is a growing urgency for these approaches because of the rapid acceleration in new coronavirus literature, making it difficult for the medical research community to keep up.
You can read more about it here
Discovery of novel antibiotic Halicin using deep learning
A recent paper has caught a lot of attention recently "A Deep Learning Approach to Antibiotic Discovery" DOI from Regina Barzilay's group at MIT. They used a deep neural network model to predict growth inhibition of Escherichia coli using a collection of 2,335 molecules, the molecules were described using Morgan fingerprints, computed using RDKit, for each molecule using a radius of 2 and 2048-bit fingerprint vectors. Using this methodology they identified the known c-Jun N-terminal kinase inhibitor SU3327 which they renamed Halicin. A quick search using MolSeeker allowed identification of the structure and inChiKey.
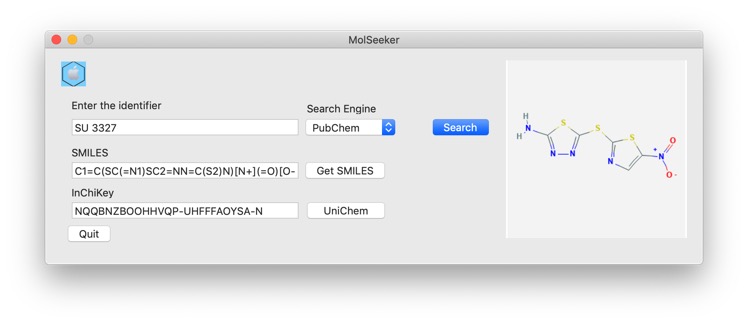
A search of UniChem using the InChikey NQQBNZBOOHHVQP-UHFFFAOYSA-N identified a number of other identifiers in different databases.
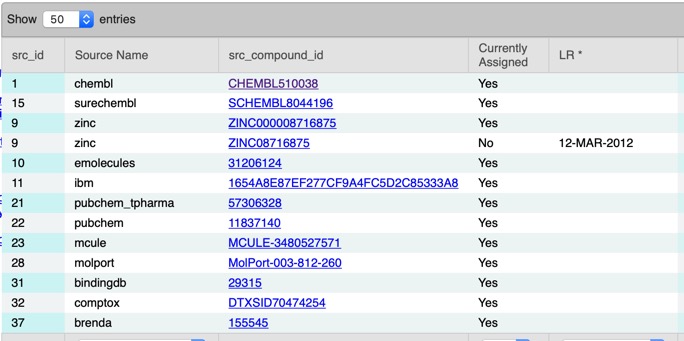
Including a link to the ChEMBL entry CHEMBL510038 giving the biological data 0.7 nM Inhibition of c-Jun N-terminal kinase by time-resolved FRET assay, and links to the original 2009 publication DOI describing the c-JNK SAR. The compound has a rat half-life of 0.45 h. There is another publication that might be of interest describing "Discovery of 2-(5-nitrothiazol-2-ylthio)benzo[d]thiazoles as novel c-Jun N-terminal kinase inhibitors" DOI.
Certainly an interesting approach, I suspect the nitrothiazole functionality would set off a few structural alerts but there are certainly of plenty of similar compounds commercially available that would allow exploration of the SAR without too much investment in resources.
All code and data is available on GitHub and there is also a website where you can test your own molecules http://chemprop.csail.mit.edu.
Upcoming Conferences
I just thought I'd mention a couple of meetings I'm helping to organise.
2nd RSC-BMCS / RSC-CICAG Artificial Intelligence in Chemistry
Artificial Intelligence is presently experiencing a renaissance in development of new methods and practical applications to ongoing challenges in Chemistry. Following the success of the inaugural “Artificial Intelligence in Chemistry” meeting in 2018 a second meeting has been organised at Fitzwilliam College, Cambridge (2nd to 3rd September 2019). The lineup is now finalised and looks like a great selection of speakers. There is still time to submit posters (closing date 5th July).
Registration is open and there are discounts for RSC members.
The Twitter hashtag - #AIChem19 is already being actively used.
20th SCI/RSC Medicinal Chemistry Symposium
This is Europe’s premier biennial Medicinal Chemistry event, focussing on first disclosures and new strategies in Medicinal Chemistry. It takes place a Churchill College, Cambridge UK, 8 September - 11 September 2019. There is a fantastic lineup of speakers and looks to be one of the highlights of the MedChem calendar. Early career scientists can also take part in a Medicinal chemistry workshop on the Sunday afternoon, a great way for people to learn medicinal chemistry and meet other scientists in a fun and informal setting.
You can register here both RSC and SCI members get a reduced rate, and despite the slightly confusing page on the SCI website you don't have to be a member to attend, just select "Event Member FREE from the dropdown menu and you can register for the event without membership.
Twenty Years of the Rule of Five
It has been over twenty years since Lipinski published his work determining the properties of drug molecules associated with good solubility and permeability. Since then, there have been a number of additions and expansions to these “rules”. There has also been keen interest in the application of these guidelines in the drug discovery process and how these apply to new emerging chemical structures such as macrocycles.
This meeting aims to have a look at the impact the Ro5 has had on drug discovery and as well as looking to the future and how we use these rules in the changing drug compound landscape as drug discovery moves into novel areas of chemistry.
There is a very exciting group of speakers and the timetable has been designed to allow a panel discussion after each session. Given the topic and the speakers I'm sure these will be entertaining sessions.
You can register here and there are discounts for RSC members
Twitter hashtag - #RuleofFive2019
2nd RSC-BMCS / RSC-CICAG Artificial Intelligence in Chemistry
The lineup for the 2nd RSC-BMCS / RSC-CICAG Artificial Intelligence in Chemistry Monday-Tuesday, 2nd to 3rd September 2019 Fitzwilliam College, Cambridge, UK has been updated.
Twitter #AIChem19
Artificial Intelligence is presently experiencing a renaissance in development of new methods and practical applications to ongoing challenges in Chemistry. Following the success of the inaugural “Artificial Intelligence in Chemistry” meeting in 2018, we are pleased to announce that the Biological & Medicinal Chemistry Sector (BMCS) and Chemical Information & Computer Applications Group (CICAG) of the Royal Society of Chemistry are once again organising a conference to present the current efforts in applying these new methods. The meeting will be held over two days and will combine aspects of artificial intelligence and deep machine learning methods to applications in chemistry.
Speakers
Deep learning applied to ligand-based de novo design: a real-life lead optimization case study, Quentin Perron, IKTOS, USA
A Turing test for molecular generators, Jacob Bush, GlaxoSmithKline, UK
Presentation title to be confirmed, Keynote: Regina Barzilay, Massachusetts Institute of Technology, USA
Artificial intelligence for predicting molecular Electrostatic Potentials (ESPs): a step towards developing ESP-guided knowledge-based scoring functions, Prakash Rathi, Astex Pharmaceuticals, UK
Molecular transformer for chemical reaction prediction and uncertainty estimation, Alpha Lee, University of Cambridge, UK
Drug discovery disrupted - quantum physics meets machine learning, Noor Shaker, GTN, UK
Presentation title to be confirmed, Christian Tyrchan, AstraZeneca,
Presentation title to be confirmed, Anthony Nicholls, OpenEye Scientific Software, USA
Deep generative models for 3D compound design from fragment screens, Fergus Imrie, University of Oxford, UK
DeeplyTough: learning to structurally compare protein binding sites, Joshua Meyers, BenevolentAI, UK
Presentation title to be confirmed, Maciej Haranczyk, IMDEA, Spain
Deep learning for drug discovery, Keynote: David Koes, University of Pittsburgh, USA
Presentation title to be confirmed, Olexandr Isayev, University of North Carolina at Chapel Hill, USA
Dreaming functional molecules with generative ML models, Christoph Kreisbeck, Kebotix, USA
Presentation title to be confirmed, Keynote: Adrian Roitberg, University of Florida, USA
Applications for poster presentations are welcomed, the closing date for submission is 5th July. A number of RSC-BMCS and RSC-CICAG student bursaries are available up to a value of £250, to support registration, travel and accommodation costs for PhD and post-doctoral applicants studying at European academic institutions. The closing date for bursary applications is 15th July.
Full details are on the conference website
Atomwise AIMS awards

I suspect many will have noticed the recent announcement of the Early Results in Drug Discovery Partnership with AI Biotech Company. These are the first results of the Atomwise AIMS awards:
The researchers have been using Atomwise’s AI-powered in silico screening technology to develop therapeutic treatments for, among others, certain types of strokes, hand-foot-and-mouth disease, and an infection that causes reproductive failure in pigs.
The AIMS award program is a great opportunity for university research scientists to easily access AI-assisted structure-based virtual screening technology:
- Customized small molecule virtual screen using AtomNet™ technology
- 72 small molecules predicted to bind to a specific target protein – QC verified by mass spectrophotometry, resuspended and diluted to a convenient concentration, aliquoted into microtiter plates, and delivered at no cost to the researcher
- Support from Atomwise’s medicinal chemists and structural biologists
- Opportunity to receive up to $30K USD to subsidize assay work
If you have a target protein with an X-ray crystal, Cryo-EM, or NMR structure, or with close sequence homology to a protein with available structures, and an assay in place to evaluate 72 potential hits, then you should consider applying.
Full details are on the AIMs awards page and the closing date is 29 April 2019.
Encouraging early results for the drug delaying onset of Motor Neurone discovered by artificial intelligence
Motor neurone disease (MND) describes a group of diseases that affect the nerves (motor neurones) in the brain and spinal cord, is is likely that there are multiple molecular targets. Amyotrophic lateral sclerosis (ALS) also known as Lou Gehrig's disease is the most common form of MND. Edaravone was recently approved for the treatment of ALS but the mechanism is unknown. It is a free radical scavenger and oxidative stress has been hypothesised to be part of the process that kills neurones in people with ALS. However new treatments are urgently needed.
For this reason I was particularly interested to read about a potential novel treatment for ALS arising from work between Benevolnet.ai and Sheffield Institute for Translational Neuroscience.
The study, led by Dr. Richard Mead and Dr. Laura Ferraiuolo at SITraN, assessed the efficacy of a drug candidate proposed by BenevolentAI's artificial Intelligence technology for Motor Neuron Disease (MND), also known as Amyotrophic Lateral Sclerosis (ALS). SITraN found there are significant and reproducible indications that the drug prevents the death of motor neurones in patient cell models, and delayed the onset of the disease in the gold standard model of ALS…Dr. Richard Mead of SITraN commented: "This is an exciting development in our research for a treatment for ALS. BenevolentAI came to us with some newly identified compounds discovered by their technology - two of which were new to us in the field and, following this research, are now looking very promising. Our plan now is to conduct further detailed testing and continue to quickly progress towards a potential treatment for ALS."
SITraN expect to publish an abstract at the Motor Neurone Disease Association 28th International Symposium in Boston in December 2017.