ABCB1 also known as P-gp and MDR1
Transporter mediated efflux of drugs is major issue in drug discovery. The best understood of the transporters is ABCB1 also known as P-gp, MDR1 (mdr1 in rodents) this transporter is present in the blood-brain barrier, in the gut, on hepatocytes, and the kidney. It is also one of the transporters found to be up-regulated in some drug-resistant tumors and is considered to be one of the major causes of treatment failure. Human isoform MDR1 (ABCB1) Mouse isoforms mdr1a and mdr1b, mdr1a predominant form in the BBB and intestine in mouse, mdr1b and mdr1a expressed in kidney and liver.
P-gp is a 170-kDa glycoprotein that is encoded by the MDR1 gene. A low resolution crystal structure of mouse P-gp has been described by Aller et al (The PDB code is 3G5U). The protein consists of two homologous domains each containing 6 transmembrane spanning domains with a nucleotide binding domain connected by a short linker. Click here to view in JMOL in a new window. Each domain contains 6 helical domains thought to be transmembrane spanning. The x-ray structure of apo P-gp at 3.8 angstroms reveals an internal cavity of approximately 6000 angstroms3 with a 30 angstrom separation of the two nucleotide-binding domains. Two additional P-gp structures (3G60 and 3G61) with cyclic peptide inhibitors demonstrate distinct drug-binding sites in the internal cavity. Two charged residues are present in the cavity of Pgp, a Glu and His, which may be relevant for the binding of the basic substrates. Pgp recognizes a wide variety of structurally diverse substrates and it is thought substrate interactions occur within the inner leaflet of the bilayer thus membrane partitioning is essential for Pgp interaction subsequent drug transport entails ATP hydrolysis and large conformational changes to the protein.
The diversity of substrates susceptible to P-gp mediated efflux is well noted, with a wide variety of chemotypes shown to be substrates spanning a range of therapeutic classes. It is possible to evalauate compopunds in permeability assays. In these types of studies, permeability studies in the apical-to-basolateral and in the basolateral-to-apical direction are performed and the ratio of these numbers can then be used to provide some clues about P-gp involvement.
MDCK-hMDR1
Canine kidney cells transfected with hMDR1 gene, also used as model of blood brain barrier because of high level PGP expression. Measure permeability both ways across membrane A-B and B-A.
- Papp > 150 nm/s high permeability
- Papp 50-150 Medium
- Papp < 50 nm/sec Low
- BA/AB < 2 no significant efflux
- BA/AB 2-5 modest
- BA/AB > 5 Extensive efflux
- Can use Cyclosporin as PGP inhibitor to confirm PGP substrates.
The APTPase activity of P-gp is closely correlated with the transport activity and measurement of the ATPase activity has been used as a marker for P-gp substrate activity. The generation of P-gp knockout mice has also enabled a better understanding of the role of P-gp in drug distribution. There is a table of known PGP substrates (and inhibitors) here http://www.genemedrx.com/PGPtable.php

The correlation between the in vitro data using the mouse mdr1a and the data from the PGP knockout mouse suggests the in vitro models are reasonably predictive however there is at present insufficient data to determine the reliability of the MDCK-hMDR1 for predicting in vivo effects in man.
Didziapetri et al (Journal of Drug Targeting, Volume 11, Issue 7 August 2003, 391 - 406) have built a classification model, the classification rules are based on the following factors: (i) compound's size expressed through molar weight or volume, (ii) H-accepting given by the Abraham's β (that can be crudely approximated by the sum of O and N atoms), and (iii) ionization given by the acid and base pKa values. Very roughly, SPGP can be estimated by the ”rule of fours”. Compounds with (N+O)≥8, MW>400 and acid pKa > 4 are likely to be Pgp substrates whereas compounds with (N+O)‚≤4, MW<400 and base pKa<8 are likely to be non-substrates.
There have been a number of attempts to build SAR models, however the possibility of multiple binding sites has limited the design of general pharmacophore models. Garrigues et al have described the characterization to two pharmacophores (Mol Pharmacol 62:1288-1298, 2002) using a series of cyclic peptides together with the known substrates Verapamil, Vinblastine, Progesterone. This study identified an aromatic binding region with a number of additional lipophilic binding pockets. Ekins et al (Mol Pharmacol 61:974-981, 2002) used 16 Verapamil analogues to build a pharmacophore consisting of one hydrogen bond acceptor, one ring aromatic feature, and two hydrophobes. More recently Li et al (J. Chem. Inf. Model., 2007, 47 (6), pp 2429-2438) have built a classification model using nine distinct pharmacophores. Given the mutiple binding sites this type of ensemble model may have advantages.
Jerome Hochmann has described work by the Merck group directed towards building structure-transport model using a variety of chemical descriptors. The increasing the number of hydrogen bond acceptors (HBA) and SlogP were found to have a significant influence. They were also able to identify functional groups that appeared regularly to give PGP activity, many appear to have multiple HBA.

They also suggest structural changes that might lower PGP efflux, these include reducing the pKa of amines, reducing the number of potential hydrogen bonding interactions, either by removing heteroatoms or increasing steric hindrance.
Also of note is the fact that P-gp is inducible by PXR, pregnane X receptor. PXR activation has also been shown to induce CYP2B, CYP3A, UGT1A1 and CYP2C8. It has been estimated that unwanted activation of PXR is responsible for 60% of all drug-drug interactions.
The influence of fluorine's impact on pKa and in vitro PGP-mediated efflux for a series of PDE9 inhibitors (below) has been explored DOI. Introduction of fluorine impacted both the basic amine but also the acidity of the lactam. Comparisons through the series of compounds and their fluorinated analogues reveals a systematic decrease in acidic pKa between 1.2–1.7 (pKa (acidic)), and shifts for the basic pyrrolidines between 1.1–3.3 units (pka basic). Introduction of F into the NH lactams often served to increase PGP activity, in contrast introduction into the N-Methyl lactams reduced PGP activity.
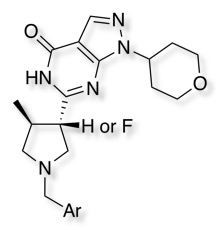
Down regulating PGP expression
An interesting publication by Kim and Bynoe in J Clin Invest DOI show that activation of the A2A adenosine receptor (AR) with an FDA-approved A2A AR agonist (Lexiscan) rapidly and potently decreased P-gp expression and function in a time-dependent and reversible manner.
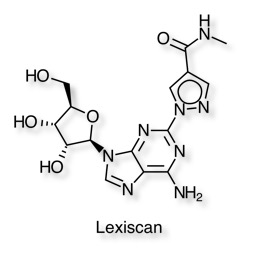
They demonstrate that down modulation of P-gp expression and function coincided with chemotherapeutic drug accumulation in brains of WT mice and in primary mouse and human brain endothelial cells, which serve as in vitro BBB models.
Worth looking at the Human Membrane Transporter Database.
Updated 2 June 2018